
ISSN 1666-7948
Revista Electrónica de Ciencia y Educación
Departamento de Química Biológica FCEN-UBA IQUIBICEN
ISSN 1666-7948
Revista Electrónica de Ciencia y Educación
Departamento de Química Biológica FCEN-UBA IQUIBICEN
Enfermedades Poco Frecuentes: De ALAPA, De los Pacientes y sus Familias para estudiantes, investigadores/as y profesores/as
Verónica Alonso, Florencia Braga Menéndez, Johana Bauer, Fabián Crespo, Georgina Davis, Alexis Descourviers, Carolina Oliveto, Vanina Sánchez, Javier Santos y Natalia Torres
ALAPA
Conflictos de intereses
Declaramos no tener ningún conflicto de intereses, no responder a ningún poder gubernamental ni actual ni pasado, no responder a ningún laboratorio o industria farmacéutica. Sí nos moviliza buscar soluciones para los/las pacientes y sus familias. Sí creemos que las soluciones son siempre inclusivas. Sí creemos que la salud es un derecho humano.
Las Enfermedades poco Frecuentes o Raras
Existen cerca de 8000 enfermedades poco frecuentes (EpoF o EFP). Cada una puede tener una prevalencia muy baja (<1 en 2000); sin embargo, todos los afectados suman ~6% de la población mundial [1]. La mayor parte de estas enfermedades aún no tienen tratamiento o cura; muchas de ellas han sido poco o nada estudiadas. Unas pocas tienen soluciones terapéuticas, pero suelen ser muy costosas para los sistemas de salud. Se ha propuesto el uso de técnicas de medicina de precisión o personalizada como una alternativa para encontrar terapias potenciales en estas enfermedades [2]. Las EpoF sin lugar a duda deberían ser un tema prioritario en la agenda de las políticas socioeconómicas y científicas.
Sin embargo, las EpoF quedan en general subrepresentadas entre los temas investigados por la comunidad científica. Asimismo, para los Estados las EpoF representan un desafío siendo la accesibilidad a los medicamentos y terapias (entre ellas las terapias génicas) un problema a escala mundial en el que está en juego el derecho humano a la Salud.
La Odisea del Diagnóstico
Uno de los primeros problemas con los que nos encontramos es el diagnóstico de las EpoF [3]. En una gran cantidad de casos el diagnóstico extremadamente lento puede llevar años y muchos profesionales involucrados; las razones de esto son numerosas, pero entre las más comunes se encuentran el desconocimiento por parte de los médicos de numerosos aspectos de la bioquímica y la medicina asociada a estas enfermedades, la dificultad en el acceso al diagnóstico molecular, la dificultad en la interpretación del diagnóstico molecular debido a la ausencia de información sobre variantes patogénicas, la dificultad del trabajo cooperativo y en redes). En los últimos años se han realizado avances significativos en lo que concierne al diagnóstico molecular y al uso de nuevas herramientas computacionales de apoyo que representan alternativas valiosas.
Los Pacientes, las Asociaciones y las Alianzas
Es cierto que el principal motor a nivel internacional para garantizar la continuidad en el estudio de EpoF han sido las asociaciones de pacientes y familiares de pacientes que empujan de manera sostenida y que han buscado apoyo y se han hecho escuchar. También es cierto que cada asociación transita por un camino que aunque desde la visión más científica es específica y requiere conocimiento enfocado, en cierta manera es también muy similar al que recorrerán otras asociaciones en todo aquello que involucre la interacción con los organismos del estado, la búsqueda de recursos y ayuda, la implementación de estrategias de diagnóstico superadoras, la organización, la generación de plataformas informativas, la accesibilidad a los medicamentos y a especialistas, y la interacción con el sistema científico tecnológico, por este motivo es que resulta extremadamente positiva la cooperación entre asociaciones de diferentes EpoF en el formato de alianzas. A esto lo podemos pensar como una estrategia útil para la transferencia eficiente de experiencia y conocimiento. ALAPA es la alianza argentina de pacientes y agrupa a muchas asociaciones de pacientes acercando soluciones y estrategias comunes que permiten2 afrontar mucho más rápidamente y en forma concertada y colaborativa el desafío que tiene por delante una asociación de una EpoF particular (Figura 1)
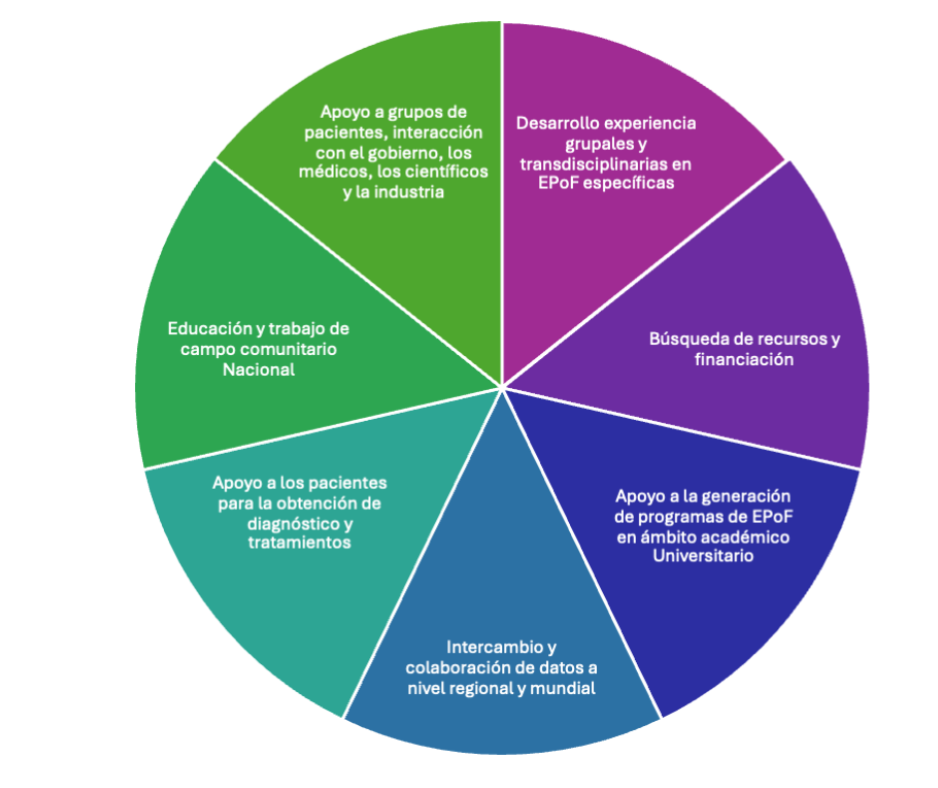
Figura 1: Algunos de los desafíos que enfrentamos los pacientes con enfermedades raras, y sus familias. Los roles de los grupos y asociaciones de pacientes, médicos, investigadores/as en EpoF, la industria y los gobiernos están interrelacionados. El intercambio de datos y la colaboración son aspectos críticos; inclusión, educación y trabajo comunitario son aspectos críticos en el campo de las EPoF a nivel Nacional.
La Alianza Argentina de Pacientes y la interacción con investigadoras/es y estudiantes
Una de las misiones más importantes de ALAPA es ayudar a promover la investigación científica traslacional, orientada a resolver problemas relevantes en el campo de la biomedicina, investigaciones preclínicas (y clínicas) y la búsqueda de estrategias para su implementación en la práctica clínica, para acercar a los pacientes al corto, mediano, o largo plazo, soluciones de diagnóstico y terapéuticas.
Desde ALAPA se busca tender lazos y puentes entre investigadores/as científicos/as y asociaciones y líderes de pacientes. Estos primeros lazos promueven el intercambio de conocimiento en una forma transdisciplinar enriqueciendo a las partes. Este es el primer paso en la generación de articulaciones y asistencia de expertos y expertas comprometidos/as con la búsqueda de conocimiento y la posibilidad de dar respuestas a problemas tan complejos. El vínculo humano de los/las investigadores/as con los pacientes, sus familias y las asociaciones humaniza a las investigaciones científicas y les da un sentido aún mucho más profundo.
Desde un punto de vista práctico, el vínculo de las asociaciones con investigadores/as especialistas permite un acceso mucho más rápido y seguro a información relevante como lo es la concerniente a medicamentos aprobados y a sus mecanismos de acción a nivel molecular. Toda esta información es útil para los pacientes y para sus familias, y también permite ganar en un nivel de comprensión más amplio.
Específicamente, la generación de proyectos conjuntos entre asociaciones e investigadores/as permite avanzar en objetivos mucho más concretos, dando continuidad, aumentar la chance de conseguir financiamiento y recursos para investigar en estos aspectos. También es posible llevar adelante trabajos en red que incluyen a grupos de investigación tanto nacionales como internacionales y mantener reuniones científicas específicas periódicas. La interacción de las asociaciones de pacientes con la Universidad permite la inclusión de nuevos contenidos, cursos y asignaturas de grado y postgrado, con contenido relevante que permite transmitir conocimientos, y despertar vocaciones en estudiantes, docentes auxiliares y profesores. La articulación de becas de grado específicamente dedicadas a estudiantes para desarrollar proyectos y dar sus primeros pasos en investigación científica en EPoF permite reforzar esas capacidades y apoyar esas vocaciones. Desde 2021, en interacción primero con el iB3 y luego con toda la FCEN, venimos promoviendo y apoyando el Programa de Becas con un total de 20 becas otorgadas hasta la fecha, obteniendo apoyo de empresas como Pfizer, y Biogen, y de asociaciones de pacientes como el Grupo Argentino Anemia de Fanconi.
Algunos ejemplos del tipo de interacciones que ALAPA están llevando adelante con el sistema de CyT incluyen a proyectos científicos en Síndrome de Rett, Atrofia muscular Espinal, Glioma difuso, Anemia de Fanconi, Discinesia Ciliar Primaria y Ataxia de Friedreich. A continuación se pueden encontrar resúmenes del estado de avance de cada proyecto. Algunos de ellos son de más reciente comienzo. Sin embargo, creemos que podría ser muy útil para los/las lectores/as y esécialmente para las y los estudiantes universitarios disponer del espectro completo.
Las asociaciones de pacientes en ALAPA, han demostrado que la colaboración y el activismo pueden tener un impacto significativo en la investigación y el tratamiento de las patologías. A través de sus esfuerzos, han logrado conectar a investigadores, formar redes de investigación y explorar estrategias de investigación. Es vital seguir apoyando estas iniciativas para acelerar el diagnóstico correcto, el tratamiento efectivo, y permitir una mejora en la calidad de vida de los pacientes afectados por estas enfermedades que muchas veces resultan devastadoras.
Atrofia muscular espinal (ORPHA:70)
Asociación en Argentina: Familias AME Argentina
Contacto: fameargentina@gmail.com
Título del proyecto: Investigación de un papel putativo para la estructura de la cromatina en la terapia génica de la atrofia muscular espinal a través del control del empalme alternativo
Integrantes: Doctor Alberto R. Kornblihtt y colaboradores.
Filiaciones: Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE-UBACONICET) Departamento de Fisiología, Biología Molecular y Celular. Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Argentina
La atrofia muscular espinal (AMS) es un trastorno genético de las neuronas motoras. La AME es la principal causa genética de mortalidad infantil y es causada por mutaciones de pérdida de función del gen SMN1 (neurona motora de supervivencia 1). En individuos sanos, SMN1, de posición telomérica, codifica la supervivencia de la proteína de la neurona motora (SMN), que es un componente importante de la maquinaria de empalme y tiene un papel crucial en el sistema nervioso. Los humanos tienen un paralogo de SMN1, llamado SMN2, de posición centromérica. Debido a una variación en un solo nucleótido, el exón 7 de SMN2 está mal incluido en el ARNm maduro, lo que hace que solo el 10% de las transcripciones de SMN2 codifiquen la proteína completamente funcional con el 90% restante codificando una proteína truncada y no funcional. Por esta razón, en ausencia de expresión de SMN1, SMN2 no puede compensar la deficiencia en la proteína SMN.
En la actualidad, la terapia más prometedora para la AME ha sido desarrollada por el Dr. Adrian Krainer del Laboratorio Cold Spring Harbor en Long Island, Nueva York. El objetivo de esta terapia es restaurar los niveles normales de expresión de SMN mediante el uso de oligonucleótidos antisentido (ASO) diseñados para aumentar la inclusión del exón 7 en la transcripción de SMN2. Krainer diseñó un potente ASO modificado con una 2'-Ometoxietilribosa (MOE) que promueve la inclusión del exón 7 en el transcrito SMN2 en células cultivadas e incluso en ratones modelo para SMA, cuando se administra sistémicamente [4]. El mecanismo molecular de la estrategia ASO modificada opera a través de la interacción del ASO con una secuencia ubicada en el intron que sigue al exón 7 (intrón 7), actuando como un silenciador de empalme intrónico (ISS) que es el sitio de unión de una proteína nuclear llamada hnRNP A1. Cuando el sitio de unión a hnRNP A1 se bloquea con el ASO modificado, la inclusión del exón 7 en el transcripto maduro aumenta y, en consecuencia, se producen niveles más altos de proteína SMN sana y de longitud completa.
La administración sistémica de la ASO en ratones modelo adultos corrigió defectos en el hígado y los riñones, pero no en la médula espinal [4]. El grupo Krainer también ha infundido el ASO en el ventrículo lateral del cerebro en ratones transgénicos adultos que expresan un transgénico SMN2 humano utilizando una micro bomba osmótica. Este tratamiento aumentó significativamente la inclusión del exón 7 de SMN2, tanto en los niveles de ARNm como de proteínas [5]. Sin embargo, cuando Krainer y sus colegas inocularon la ASO sistemáticamente en ratones recién nacidos, se obtuvo una restauración más eficiente de los niveles de SMN en el sistema nervioso, con un aumento concomitante de 25 veces en la esperanza de vida [6]. Los resultados anteriores sugirieron que la restauración de SMN en los tejidos periféricos es importante para corregir los defectos en las neuronas motoras . Además, los investigadores demostraron que un aumento de los niveles de SMN en los tejidos periféricos de los ratones neonatales era suficiente para aumentar la vida útil y mitigar los defectos característicos del SMA.
Uno de los aspectos más destacados e intrigantes de la terapia ASO ensayada en ratones es la durabilidad del efecto. Esto no se esperaría si el ASO solo interactuara con el pre-ARNm para bloquear el efecto de hnRNP A1. Por esta razón, Krainer y sus colegas sugirieron que tal5 vez el ASO también esté trabajando a nivel de ADN SMN2 modificando la estructura de la cromatina cerca del exón 7. Los cambios en la estructura de la cromatina tienden a ser más estables que los que ocurren en el ARNm, y en algunos casos se ha informado que son heredados mitóticamente.
Nuestro grupo tiene una vasta experiencia en la regulación del empalme alternativo a través de cambios en la estructura de la cromatina. Nos gustaría investigar los posibles roles de los cambios en la estructura de la cromatina en la modulación del empalme alternativo de SMN2 exón 7 por el ASO diseñado por Krainer y otros tratamientos de oligonucleótidos. En el núcleo, la molécula de ADN se asocia con proteínas llamadas histonas para formar estructuras en forma de tambor conocidas como nucleosomas. Los nucleosomas forman las unidades de repetición fundamental de la cromatina eucariota que sirve para empaquetar los grandes genomas eucariotas en el núcleo mientras se asegura el acceso apropiado a él. Los nucleosomas están organizados como una fibra de "cuentas sobre una cuerda", conectadas por tramos de ADN desnudo libre de histona, conocido como ADN de enlace. Si el ADN está más bien empaquetado, formando lo que comúnmente se conoce como heterocromatina, las "cuentas" se acercan e impiden el progreso de la enzima a cargo de copiar el ADN en el ARN (ARN polimerasa II o Pol II). El efecto opuesto ocurre cuando las "perlas" se alejan entre sí, formando eucromatina. En ese escenario, el ADN está más suelto y la enzima que hace que el ARN pueda progresar de manera más eficiente.
Nuestro grupo ha demostrado que el estado de compactación de la cromatina alrededor de la región donde se coloca un exón alternativo tiene un impacto directo en su inclusión en el ARN mensajero maduro. Los tratamientos o condiciones que promueven la apertura de la cromatina causan el salto del exón alternativo porque la enzima que produce el ARN puede progresar más rápido [7]. Por el contrario, los tratamientos o condiciones que hacen que el envase de la cromatina sea más apretado generalmente promueven la inclusión de exones alternativos porque la enzima que hace que el ARN progrese más lentamente [8]. Esta situación depende de la secuencia del exón alternativo involucrado. Hay ciertos tipos de exones que se comportan de manera opuesta a la manera explicada anteriormente, con mayor inclusión cuando la cromatina está abierta y la transcripción es rápida, y mayor salto cuando la transcripción es lenta porque la cromatina es más compacta.
Nuestro laboratorio también demostró que la introducción en células vivas de pequeños ARN interferentes (siRNA) que interactúan con secuencias de intron ubicadas en exones alternativos cercanos, modifican el patrón de inclusión de esos exones en el ARNm al inducir cambios en la condensación de la cromatina y la velocidad de transcripción [9, 10], descubriendo una nueva forma de controlar el empalme alternativo.
En este proyecto queremos investigar si la inclusión del exón 7 del gen SMN2 está regulada por la velocidad de la enzima que produce ARN a partir del ADN. Simultáneamente, investigaremos si la ASO desarrollada por el Dr. Krainer y siRNA diseñados por nuestro laboratorio cambian el patrón de empalme del exón 7 a través de cambios en la estructura de la cromatina. Nuestro laboratorio tiene las habilidades teóricas y prácticas y la experiencia necesarias para probar estas hipótesis. No estamos proporcionando aquí detalles experimentales debido a su complejidad. La Figura 2 muestra la estrategia general de la investigación propuesta. También planeamos usar medicamentos que modulan la condensación de la cromatina. Estos medicamentos podrían cambiar los niveles de inclusión del exón 7 y eventualmente usarse en combinación con la ASO y/o los siRNA en protocolos de tratamiento experimental.
Este proyecto es una colaboración entre los laboratorios Krainer y Kornblihtt con financiación de Familias AME Argentina. Las ASO y otras herramientas moleculares específicas serán proporcionadas por el laboratorio Krainer, pero la mayor parte del trabajo de laboratorio será realizado en nuestro laboratorio en Buenos Aires por el estudiante de doctorado Luciano Marasco, que estará bajo mi supervisión directa. Discutiremos los resultados con el Dr. Krainer semanal. Si es necesario, Luciano puede viajar al Dr. Laboratorio de Krainer en Long Island. El salario de Luciano estará cubierto por una beca de CONICET. Luciano tiene tres años de experiencia trabajando en nuestro laboratorio, en temas relacionados fue coautor de un artículo publicado en la prestigiosa revista Molecular Cell [11].
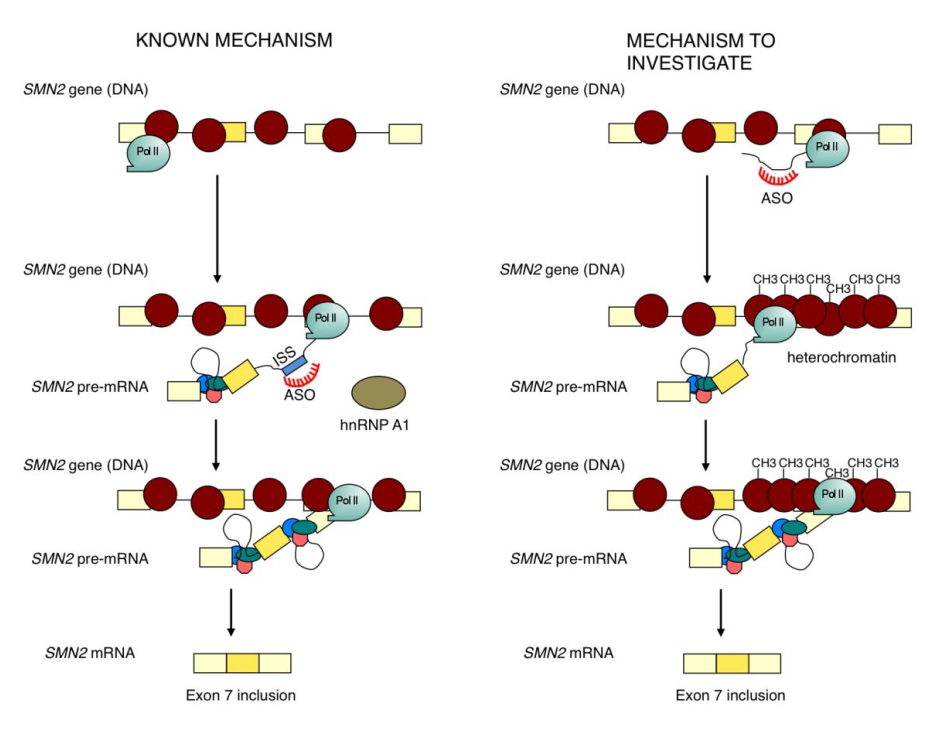
Figura 2: En el modelo actual de la regulación de SMN2 exon 7 empalme de la estructura de cromatina (izquierda) no juega ningún papel. Tras el tratamiento con ASO, los nucleosomas (círculos rojos) no cambian su distribución a lo largo del gen SMN2. El ASO actúa bloqueando la unión de la proteína reguladora negativa hnRNP A1 a la ISS presente en el ARNm, lo que promueve la inclusión del exón 7 en el ARNm maduro. A la derecha, el ASO se une al ARN sintetizado por la ARN polimerasa II (Pol II). Esta enzima funciona de izquierda a derecha en el esquema, sintetizando ARN utilizando ADN como plantilla. La unión de la ASO al ARN promueve una condensación de los nucleosomas, generando una región local de heterocromatina. Esta condensación es la consecuencia de la adición de grupos metilo a las histonas. La cromatina condensada actúa bloqueando la progresión de Pol II, lo que a su vez causa una mayor inclusión del exón 7 en el ARNm maduro.
Anemia de Fanconi (ORPHA:84)
Asociación en Argentina: Grupo Argentino Anemia de Fanconi
Contacto: anemiadefanconiargentina@gmail.com
Título del Proyecto: Producción de una variante troyana de FANCF de longitud completa recombinante en E. coli
Etapa en la que se encuentra el proyecto: iniciado.
Necesidad de financiamiento: Si. Cuenta con una beca de grado financiada por el Grupo Argentino Anemia de Fanconi.
Modalidad: transdisciplinar
Integrantes: Michelle N. Guarino1, Cecilia Córdoba2, Carolina Oliveto2, María Florencia Pignataro1,3, Sara Frías Vázquez4 y Javier Santos1,2,3
Filiaciones: 1 Instituto de Biociencias, Biotecnología y Biología Traslacional (iB3). Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. 2ALAPA, 3CONICET. Argentina. 4Universidad Nacional Autónoma de México.
La anemia de Fanconi es una enfermedad poco frecuente (1:350.000) autosómica recesiva en la mayor parte de los casos. Desde un punto de vista molecular, más de veinte proteínas que forman parte de la vía FA/BRCA mantienen la integridad del genoma, promoviendo su reparación mediante la eliminación de cross-links entre cadenas del DNA. Las variantes patogénicas de estas proteínas resultan en una alteración del sistema de reparacio1n del DNA y como consecuencia directa en alteraciones cromosómicas. La enfermedad está caracterizada por pancitopenia progresiva (el número de glóbulos rojos, glóbulos blancos y plaquetas en la sangre es más bajo de lo normal) con insuficiencia de la médula ósea, malformaciones congénitas variables y predisposición a desarrollar tumores sólidos o hematológico. Una de las actividades enzimáticas mejor descrita de este complejo de proteínas es la de E3-ubiquitina ligasa que se realiza por las proteínas FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FAAP100, FAAP202 [12]. Además, FANCL posee un dominio RING que recluta a UBE2T (conocida también como FANCT) [13]. En particular, FANCL tiene actividad ubiquitina ligasa E3, mientras que UBE2T tiene la actividad de tipo E2. Estas actividades son necesarias para la ubiquitinación de las proteínas FANCD2 y FANCI, que intervienen en el reclutamiento de proteínas FANC “rio abajo” FANCD1, FANCJ, FANCN, FANCO, FANCP, FANCQ, FANCR, FANCS, FANCU, FANCV, FANCW, involucradas en la reparación por recombinación homóloga del DNA.
Las células de pacientes con mutaciones en genes “rio arriba” de FANCD2-FANCI de la vía FA/BRCA y que forman el complejo core, no tienen la capacidad de ubiquitinar el complejo FANCD2-FANCI, para permitir la reparación del DNA, evento crítico en el mantenimiento de la estabilidad del genoma. Un paso clave en la vía es efectivamente la monoubiquitinación de cada una de las dos subunidades (FANCI y FANCD2) del complejo ID2. La ubiquitinación de FANCD2 aumenta la afinidad del complejo por el ADN bicatenario mediante la promoción de un cambio conformacional, mientras que la ubiquitinación de FANCI protege a FANCD2 de la desubiquitinación [14], estas proteínas forman una abrazadera sobre el DNA y la ubiquitinación evita que la abrazadera se abra y se separe del DNA protegiéndolo de la degradación durante la reparación.
En este contexto molecular, FANCF y funciona como parte de la plataforma molecular sobre la que se asocian las subunidades, específicamente forma un heterotrímero con las proteínas FANCC y FANCE que es indispensable para la actividad del complejo core. Existe información estructural concerniente a un fragmento de FANCF (difracción de rayos X, PDB ID: 2iqc, [15]) y más recientemente se ha logrado determinar la estructura del complejo core (CryoEM, PDB ID: 7kzp, [16]. La estructura del complejo consta de dos copias de todas las subunidades excepto FANCC, FANCE y FANCF. Esta estequiometría parecería ser8 fundamental para su función de ubiquitina ligasa y le da un aspecto asimétrico al complejo [16]. Las variantes patogénicas que se han reportado en los pacientes AF a nivel mundial, muestran que cerca del 90% de ellas se encuentran en FANCA (60-65%, FANCC (10-15%) o FANCG (10%) y el resto presenta VP en cualquiera otro gen. En cerca del 2% de los pacientes AF la proteína FANCF se encuentra alterada. Aunque hay pocos reportes específicos sobre la falla de FANCF, lo que se ha observado es un fenotipo clínico variado e inestabilidad cromosómica típica de la AF; en dos pacientes, presentación temprana de falla medular y en uno de leucemia mieloide aguda [17, 18]. Es entonces evidente que en la vía FA/BRCA, la proteína FANCF y sus asociadas en el heterotrímero, juegan un papel fundamental para mantener la estabilidad genómica. En el caso de una paciente, se han detectado dos frameshifts (uno en cada uno de sus alelos), produciendo proteínas truncadas, una de ellas definitivamente no funcional (c.224del; p.(Gly75Valfs*6), la otra quizá con alguna funcionalidad residual (c.743_744del; p.(Phe248Cysfs*17).
En este proyecto planteamos la producción de la proteína FANCF como una variante recombinante troyana, con capacidad de ingresar a las células eucariotas, mediante la adición dela secuencia TAT en el extremo N-terminal. Una vez obtenida la proteína recombinante con un >98% de pureza la caracterizaremos conformacionalmente y evaluaremos su ingreso a células modelo (HEK293-T) mediante microscopía de fluorescencia. En una segunda etapa podremos implementar la transducción in vitro a una línea celular (obtenida pacientes) con la proteína FANCF deficiente y determinar si el fenotipo celular AF revierte, mediante estudios de inestabilidad cromosómica. Actualmente en nuestro laboratorio, estamos avanzando en el replegado de la proteína FANCF de longitud completa recombinante (Figura 3).
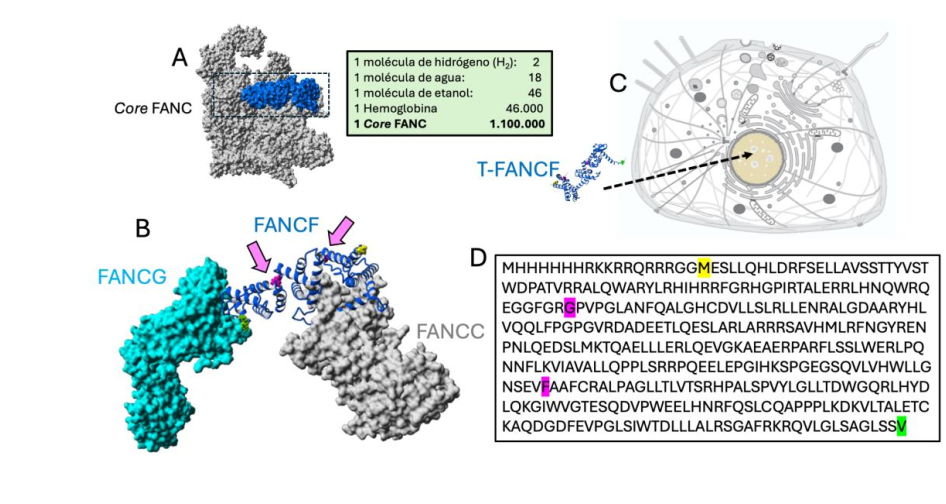
Figura 3: Rol estructural de FANCF y estrategia propuesta. (A) Localización de la subunidad FANCF (azul) en el complejo core de proteínas FANC de aproximadamente 1.100.000 Da. (B) FANCF interacciona con las subunidades FANCG y FANCC y es altamente probable que dichas interacciones le confieran estabilidad al complejo FANC. Las flechas en rosa muestran los puntos de frameshift mencionados en el texto que resultarán en la generación de sitios prematuros de terminación de la proteína derivando la ausencia del “puente estructural” FANCF. (C) Estrategia Trojana,T- FANCF tiene la capacidad de penetrar células y esta propiedad se la confiere la adición del péptido TAT de secuencia RKKRRQRRR. debe plegarse correctamente y alcanzar la localización subcelular correcta (el núcleo). (D) la secuencia de la proteína T-FANCF recombinante. En rosa se muestran los puntos de frameshift mientras que en amarillo y en verde se muestran los residios N- y C-terminal.
Ataxia de Friedreich (ORPHA:95)
Asociación en Argentina: Asociación Civil de Ataxias Argentinas (ATAR)
Contacto: ataxias@atar.org.ar
Título del Proyecto: Nanoanticuerpos troyanos para aumentar la función de la frataxina
Etapa en la que se encuentra el proyecto: avanzado
Necesidad de financiamiento: No. El proyecto cuenta con financiamiento de la Friedreich's Ataxia Research Alliance hasta Junio de 2025
Modalidad: Búsqueda de nuevas estrategias terapéuticas desde un laboratorio de estudio de proteínas.
Integrantes: María Florencia Pignataro1,3 1,3 y Javier Santos1,2,3
Filiaciones: 1Instituto de Biociencias, Biotecnología y Biología Traslacional (iB3). Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. 2ALAPA, 3CONICET. Argentina.
Los centros ferrosulfurados (Fe-S) son cofactores esenciales presentes en todas las formas de vida conocidas, y cientos de proteínas requieren tales cofactores para funcionar (Figura 4A) [19]. En las células eucariotas, la biogénesis de la mayoría de los centros Fe-S ocurre en las mitocondrias. El proceso implica las interacciones y actividades de varias proteínas clave que forman un supercomplejo, a saber, la L-Cys desulfurasa NFS1, el heterodímero APC-ISD11, la subunidad de andamiaje ISCU y el activador cinético frataxina (FXN), una proteína esencial(Figura 4B). El supercomplejo tiene una estructura heterodecamérica: (NFS1/ACP-ISD11/ISCU/FXN)2 [20]. En pacientes con ataxia de Friedreich, la expresión de FXN disminuye considerablemente o se producen variantes alteradas; algunas de estas variantes son conformacionalmente inestables [21].
Los nanoanticuerpos (NB) son proteínas de 15 kDa (Figura 4C). Muestran baja inmunogenicidad y alta estabilidad, y pueden usarse como sondas permeables específicas para objetivos tanto extracelulares como intracelulares [22]. Los NBs se están utilizando para investigar nuevas estrategias terapéuticas para diversos trastornos neurodegenerativos. Propusimos aumentar la estabilidad conformacional de variantes inestables de FXN para mejorar su función mediante la interacción NB-FXN (Figura 4D). Con este objetivo, hemos seleccionado, mediante la estrategia de presentación en fagos, varios nanocuerpos específicos. Los NBs se expresaron en bacterias E. coli, se purificaron y la interacción NB-FXN se caracterizó in vitro mediante cromatografía de exclusión por tamaño, interferometría. Los valores de KD están en el rango nanomolar de 3-30 nM. La interacción es fuerte presentando un equilibrio de disociación lento. Además, la región de la FXN implicada en la interacció (con resolución a nivel de residuos de aminoácidos) se está estudiando mediante RMN. La interacción produce una estabilización significativa de la variante patogénica G130V FXN, con un cambio de Tm más de 15 °C mayor que el mostrado por la variante G130V aislada e incluso mayor que el FXN de tipo salvaje.
La función FXN se está estudiando in vitro mediante un ensayo de activación de Cys desulfurasa NFS1, realizado en presencia de NBs. Algunos de los NBs presentan efectos inhibitorios incluso en relaciones 1:1 (NB:FXN), mientras que otros son neutrales. Para evaluar los efectos biológicos de la interacción de NB en el entorno celular, expresamos algunos de los NBs en células humanas e investigamos su direccionamiento a las mitocondrias. La expresión de algunos de los NBs no produjo alteraciones significativas de actividades enzimáticas dependientes centros Fe-S como lo son las actividades ejercidas por las enzimas aconitasa y succinato deshidrogenasa, sugiriendo que podrían ser útiles para la estabilización de variantes inestables de FXN in vivo.
En este contexto desde nuestro laboratorio planteamos la siguiente hipótesis de trabajo: los NBs intramitocondriales específicos de FXN de alta afinidad pueden modular la función de FXN de variantes inestables in vivo por lo que la administración de NBs con capacidad de penetrar células direccionados a las mitocondrias podrían representar una nueva estrategia.
Algunos de los objetivos específicos que llevamos adelante son: Preparar versiones troyanas [23] de los NB seleccionados específicos para FXN, para el delivery de proteínas. Evaluar los efectos biológicos de la expresión de NBs (y de se delivery en la versión troyana) en células de pacientes con Ataxia de Friedreich. Necesitamos investigar la viabilidad celular, el ensamblaje del grupo Fe-S, las actividades enzimáticas, y el metabolismo, la respiración celular y la sensibilidad a las especies reactivas del oxígeno. Estudiar la toxicidad y liberación de los NB en organismos. Estudiar la inmunogenicidad de los NB.
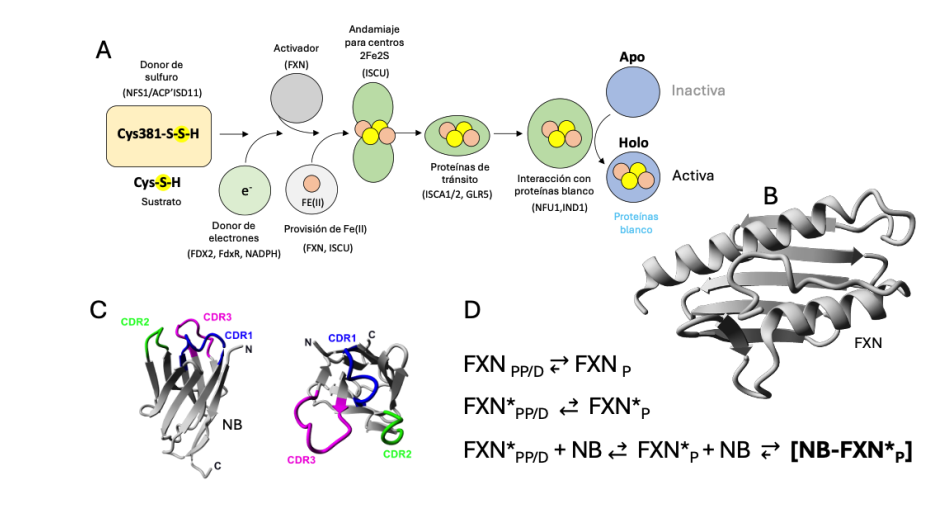
Figura 4: Nanoanticuerpos troyanos para aumentar la estabilidad de la frataxina. (A) Esquema de la vía de biosíntesis de centros FeS y su delivery a las proteínas blanco. (B) Esquema de la estructura de la FXN. Modelo de cintas. (C) Esquema de la estructura de un nanoanticuerpo (NB) mostrando en colores sus regiones variables CDR1, CDR2 y CDR3, que presumiblemente formarían el paratope de interacción con FXN. (D) Ecuaciones describiendo la hipótesis del proyecto (D, PP, y P indican estado desplegado, parcialmente plegado, y plegado, respectivamente. NB indica nanoanticuerpo. El asterisco (*) indica que se trata de una variante patogénica inestable de FXN, por eso su tendencia a desplegarse.
Glioma difuso (ORPHA:497188)
Asociación: ALAPA / Mamas y papas contra GLIOMAS
Contacto: gliomadifuso@gmail.com
Título del Proyecto: "Qué hacer cuando te dicen que ya no hay nada más que hacer? Armar un red y vincular a investigadores"; "Desarrollo de Modelos de Glioma Pediátrico Para la Investigación en Neuro-Oncología Básica y Traslacional en la Argentina."; "Implementación de un Biobanco de Modelos Tumorales y Articulacion de una Red Interdisciplinaria de Investigadores"
Etapa en la que se encuentra el proyecto: iniciado
Necesidad de financiamiento: Si.
Modalidad: transdisciplinar
Integrantes: Dr. Matías Blaustein1,6, Dra. Marianela Candolfi1,10, Dra. Hebe Alicia Duran1,2, Dr. Juan Garona1,4,5,8, Dra. Irene L. Ibañez1,2,12, Dr. Andrés Juan Kreiner1,2, Dr. Felipe Nuñez, Dr. Nicolá s Palomar, Dra. Carolina Inés Perez Castro1,7, Dr. Cesar German Prucca1,3, Dra. Natalia Rubinstein1,6, Dr. Francisco Velázquez Duarte2,6, Dr. Guillermo Agustín Videla Richardson1,9,11, Alexis Descourvieres13
Filiaciones: 1CONICET, 2CNEA, 3CIQUIBIC, 4COMTRA, 5HEC-CEMET, 6IB3, 7IBIOBAMPSP, 8IBIOCAN, 9IBYME, 10INBIOMED, 11INEU, 12UE-INN, 13ALAPA
Los gliomas pediátricos, en particular el glioma pontino intrínseco difuso (DIPG) y el glioma difuso de línea media (DMG), son tipos de cáncer cerebral altamente agresivos que afectan a niños [24]. A pesar de los avances en la medicina y la oncología en otros tipos de neoplasias, estos tumores siguen siendo intratables y tienen un pronóstico devastador, siendo actualmente la principal causa de muerte por cáncer en los niños. La falta de estrategias terapéuticas y de inversión en investigación de esta patología, a pesar de los avances en GBM de adultos, hace de la situación una emergencia. En Argentina, una asociación de padres de niños afectados por estos tumores ha tomado la iniciativa de conectar a investigadores locales y formar una red de colaboración con el objetivo de encontrar nuevas estrategias de tratamiento [25]. En esta sección se discute cómo se llevó adelante el armado de la red de investigación y las distintas estrategias de estudio, análisis, conocimiento y tratamiento de los gliomas pediátricos que pueden investigarse localmente, en nuestro País.
Contexto y Descripción de la Asociación: La asociación “Mamas y papas contra GLIOMAS”/ALAPA en Argentina surgió como una respuesta al devastador impacto de los gliomas pediátricos en sus familias. Fundada por padres de niños fallecidos, la misión de la asociación es aumentar la concienciación sobre estos tumores, proporcionar apoyo a las familias afectadas y recaudar fondos para la investigación. La asociación organiza eventos educativos y campañas de sensibilización, creando una comunidad de apoyo para las familias y un puente entre los pacientes y los investigadores.
Conectar y Vincular Investigadores: Una de las contribuciones ha sido conectar a investigadores locales que trabajan en distintos aspectos relacionados con neurooncología, invitando a que amplíen sus trabajos a los casos pediátricos, no extrapolables con los adultos por ser entidades biológicas completamente diferentes. A través del diálogo y comunicación constantes, reuniones de trabajo y la organización de jornadas, la red ha facilitado el intercambio de conocimientos y la colaboración entre científicos. Por ejemplo, la Primera Jornada Argentina de Gliomas, realizada el 14 de agosto de 2023, donde investigadores y becarios de distintas universidades argentinas y extranjeras presentaron sus estudios y discutieron posibles colaboraciones futuras. Los testimonios de investigadores destacan cómo estas conexiones han acelerado el progreso de sus investigaciones.
Formación de una Red de Investigación: La creación de la red de investigación fue un hito significativo. Esta red (CRIO, Figura 5) reune a diversas Instituciones, Universidades y laboratorios, que trabajan de manera conjunta para abordar los desafíos que presentan los gliomas pediátricos. La asociación de padres juegó y juega un papel clave, facilitando reuniones periódicas y promoviendo la participación activa de todos los miembros. Esta red no solo mejora la comunicación entre los investigadores, sino que también permite un uso más eficiente de los recursos disponibles y la búsqueda colaborativa de recursos nuevos.
Estrategias de Investigación y Terapias: Los conocimientos y capacidades de la red de investigadores en Argentina incluyen una variedad de enfoques y posibles estrategias terapéuticas innovadoras para abordar los desafíos que presentan los gliomas pediátricos, incluyendo DIPG y DMG.
Terapias Genéticas: Investigaciones sobre mutaciones específicas en DIPG y DMG, con el objetivo de desarrollar terapias dirigidas. Inmunoterapias: Estudio de cómo el sistema inmunológico puede ser entrenado para atacar las células tumorales. Tratamientos Personalizados: Uso de datos genómicos para diseñar tratamientos específicos para cada paciente. Esto incluye la secuenciación del tumor para identificar mutaciones únicas y la selección de medicamentos que apunten específicamente a esas alteraciones genéticas.
Terapias Combinadas: Investigación sobre la combinación de radioterapia y nuevos agentes quimioterapéuticos para mejorar la eficacia del tratamiento. La combinación de diferentes modalidades de tratamiento puede ayudar a superar la resistencia a un solo tipo de terapia.
BNCT (Boron Neutron Capture Therapy): Esta técnica utiliza un compuesto de boro que se acumula en las células tumorales. Cuando se irradia con neutrones, el boro produce partículas alfa que destruyen las células cancerosas desde dentro, mientras que las células normales se ven mínimamente afectadas.
Terapia Fotodinámica: Esta estrategia implica el uso de un agente fotosensibilizador que se activa mediante luz de una longitud de onda específica, generando especies reactivas de oxígeno que destruyen las células tumorales.
Plasticidad tumoral: Estudio de los mecanismos celulares y moleculares que gobiernan el proceso de plasticidad tumoral: rol de nuevos mediadores en la identidad y comportamiento celular. Los investigadores están examinando las propiedades mecánicas de las células tumorales para comprender mejor cómo estos tumores invaden el tejido cerebral. Este conocimiento puede conducir al desarrollo de nuevas terapias que alteren la mecánica celular y reduzcan la invasión tumoral.
Galectinas: Estas proteínas están involucradas en la regulación del crecimiento celulary la apoptosis, y se ha observado que están desreguladas en muchos tipos de cáncer. Los estudios están investigando cómo las galectinas pueden ser utilizadas como dianas terapéuticas para inhibir el crecimiento tumoral y promover la muerte celular.
Nanopartículas: Uso de nanopartículas para entregar fármacos directamente a las células tumorales, minimizando los efectos secundarios y mejorando la precisión del tratamiento. Además, están explorando cómo la manipulación de la elasticidad celular puede reducir la capacidad de invasión de los tumores. Creación de modelos experimentales de glioma pediátrico: hay una necesidad urgente de desarrollar modelos experimentales para mejorar la investigación y las terapias. La falta de modelos específicos ha retrasado el desarrollo de estrategias terapéuticas eficaces para avanzar en el tratamiento de esta enfermedad.
Evaluación de targets: El estudio de la participación de algunas proteínas blanco en la bioquímica de las células tumorales. Estas proteínas blanco o targets de interés por su relevancia y su asociación con cáncer son: AKT, RUNX, FOXP3, KANSL2, AHCYL1.
Objetivos Específicos:
Metodología

Figura 5: El Consorcio de Referencia en Investigación Oncopediátrica. Busca convocar profesionales, vincular instituciones, promocionar el trabajo en red, proponer opciones, conseguir fondos, incentivar, difundir lo realizado, becas de formación, facilitar la investigación científica en oncopediatría.
Sindrome de Rett (ORPHA:778)
Asociación: ALAPA / Fundación Síndrome de Rett
Contacto: argentinasinrett@gmail.com
Es un desorden del neurodesarrollo de base genética que afecta la gran mayoría de las veces al sexo femenino (95/98%). Fue descubierto en 1966 por el neuropediatra Austriaco Andreas Rett, luego en 1999 la Dra, Huda Zoghbi descubrió que este conjunto de síntomas era debido a una mutación el gen MeCP2 (Figura 6) [26]. Localizado en el cromosoma X, este gen permite la expresión de la proteína MeCP2, un regulador de la transcripción de numerosos genes que ha sido relacionado con los procesos de diferenciación y maduración neuronal. En los últimos años se ha avanzado significativamente en la comprensión de los aspectos moleculares que conducen a la patología [27, 28]. Debido a su gran distribución y amplio rango de acción, las alteraciones del gen MeCP2 desencadenan una patología diversa a la par que compleja. que afecta a 1 de cada 10.000 niñas nacidas en todo el mundo. A raíz de la afectación del sistema nervioso se generan problemas en el área cognitiva, sensorial, motora, conductual, respiratoria, digestiva y cardiaca.
"Es un trastorno del neurodesarrollo y no es neurodegenerativo" ¿Como se manifiesta? En los primeros meses de vida, su desarrollo es el esperado, pero entre los 6 y los 18 meses comienzan los síntomas característicos. Si se han adquirido habilidades se produce un período de regresión o de estancamiento en el desarrollo de las mismas. Se generan pérdidas de las habilidades lingüísticas, problemas motores, problemas de coordinación y pérdida del contacto visual. La pérdida del uso funcional de las manos es seguida por movimientos repetitivos o estereotipias. Otros síntomas son: dificultad para masticar, problemas digestivos, problemas de sueño, crecimiento retardado, crisis epilépticas, escoliosis severa, apneas e hiperventilación, entre otras. Para confirmar el diagnóstico clínico se debería consultar a un neurólogo o pediatra especialista en neurodesarrollo. Lo recomendable es realizar un estudio genético.
La Asociación: Somos una Fundación, sin fines de lucro, formada por familiares de personas con Síndrome de Rett. Unidos por una misma razón, fundamos la Fundación Síndrome de Rett – “Sin Rett” – en el 2021 con el objetivo de representar a las personas que tienen este Síndrome. Nuestra misión es darle mayor visibilidad, generar conciencia e inclusión, recaudar fondos, establecer vinculación con investigadores y construir alianzas estratégicas para mejorar la calidad de vida y acercarnos a una cura.
Conectar y Vincular Investigadores: En este sentido la vinculación que se ha logrado con investigadores de diferentes áreas nos ha permitido realizar y/o colaborar en 3 proyectos en diferentes áreas que se encuentran iniciados, buscan financiamiento y tienen una modalidad transdiciplinar (se describen más abajo, en detalle). Además, nos sumamos al trabajo de investigación del Dr. David Bruque del SAMIC-Calafate, que consiste en un método más económico y rápido de secuenciación del Gen MeCP2 , lo que permitió poder brindar diagnósticos a familias de toda la Argentina. Por otro lado, la Dra. Romina Brasca esta llevando adelante el diseño y la síntesis de compuestos de coordinación del tipo rutenio-bipiridina que resulten apropiados para contener proteínas (MeCP2, entre otras), neurotransmisores y neuromoduladores de interés en el estudio y tratamiento del Síndrome de Rett y otras patologías.
Identificación de microRNAs como biomarcadores con valor diagnóstico y pronóstico de la severidad de las manifestaciones clínicas en el síndrome de Rett
Este proyecto está dirigido por el Dr. Eduardo Cánepa, Investigador Principal CONICET, Profesor Consulto UBA, Instituto de Química Biológica de Ciencias Exactas y Naturales. UBA-CONICET. ecanepa@qb.fcen.uba.ar
El objetivo del proyecto consiste en determinar el perfil de expresión global de15 microRNAs (miRNAs) en niñas y adolescentes afectadas por el síndrome de Rett y en niñas y adolescentes neurotípicas e identificar aquellos que presenten una expresión diferencial entre ambos grupos. Paralelamente evaluar correlaciones Síntomas Diagnóstico a los 0, 20, 40, 60, 80, 100 meses. LA PROPUESTA: Determinar un biomarcador de diagnóstico y de pronósticode la severidad de las manifestaciones clínicas para el síndrome de Rett significativas entre los miRNAs diferencialmente expresados con la severidad de las manifestaciones clínicas. De este modo, se podrán identificar, en primer lugar, potenciales biomarcadores para el diagnóstico temprano y, en segundo lugar, biomarcadores pronósticos de la severidad de la enfermedad. Esto permitirá aplicar intervenciones terapéuticas más tempranas y dirigidas a las niñas afectadas. Se agrega, como objetivo adicional, evaluar la expresión diferencial de miRNAs para contribuir a la comprensión de los mecanismos moleculares involucrados en las manifestaciones fenotípicas de la enfermedad. El reclutamiento de participantes se realizará a través de la base de datos de la Fundación sin Rett (Rosario, Argentina, https://fundacionsinrett.org.ar) que reúne a familiares de pacientes afectados por el síndrome de Rett. También se cuenta con la colaboración de ALAPA (Alianza Argentina de Pacientes, http://alianzapacientes.org/inicio) es una asociación civil sin fines de lucro que tiene como propósito brindar apoyo y procurar soluciones a pacientes y familias con diagnóstico de enfermedades genéticas, poco frecuentes o de difícil diagnóstico propiciando la creación de grupos de pacientes. Grupo Casos: Niñas y adolescentes entre 3 y 17 años con diagnóstico clínico de síndrome de Rett y análisis genético que indique el tipo de mutación en el gen MeCP2. La gravedad de las manifestaciones de la enfermedad será evaluada por un score de severidad clínica de los síntomas de Rett realizada por el médico que atiende a cada paciente. Grupo Control: Niñas y adolescentes sanas pareadas por edad preferentemente de la misma ciudad de residencia y de similar nivel socioeconómico. Tamaño de la muestra: Entre 40 y 50 participantes de cada uno de los grupos. Ya se tomaron el 60/65% de las muestras. El protocolo fue aprobado por el Comité de Ética en la investigación de la Sociedad Argentina de Investigación Clínica (SAIC) Código 8033.
Nuevas estrategias diagnósticas para el Síndromes de Rett”
Este proyecto está dirigido por la Dra. Marisol Delea y el Dr. Carlos David Bruque - Hospital de Alta Complejidad El Calafate S.A.M.I.C. - Unidad de Conocimiento Traslacional Hospitalaria Patagónica - CONICET. secretaria.ucthp@gmail.com En Argentina, nos enfrentamos a la dificultad de acceso a pruebas de secuenciación genética en el sistema de salud público, lo que dificulta aún más el proceso de diagnóstico para los pacientes. Dependiendo en gran medida de laboratorios privados, muchos pacientes experimentan retrasos significativos en la obtención de un diagnóstico molecular preciso.
Es fundamental abordar esta problemática y trabajar hacia soluciones que mejoren el acceso al diagnóstico genético para los pacientes con Síndrome de Rett en nuestro país. Además, la identificación de variantes puntuales del gen MeCP2 es de vital importancia no solo para el diagnóstico preciso, sino también para el desarrollo de futuros tratamientos, incluida la terapia génica y la edición genética.
Nuestro objetivo es implementar una metodología de secuenciación más eficiente y económica, utilizando tecnología de secuenciación masiva en un equipo MiSeq. Esta estrategia permitirá ampliar el alcance de la secuenciación genética, lo que resultará en una mayor capacidad para identificar variantes puntuales del gen MeCP2 en un mayor número de pacientes, y a un costo más accesible para el sistema de salud público. A la vez está estrategia desarrollada podría implementarse a otros genes relacionados con la patología que tienen variantes génicas. Ya se enviaron más de 20 muestras de todo el país y seguimos trabajando.
Punto tecnológico para la accesibilidad de personas con síndrome de Rett y otras condiciones neurológicas.
Este proyecto está dirigido por la Dra. María Andrea Guisen – Dra. en Ciencias Informáticas - Experto y Avanzado en TIC y Discapacidad – Lic. en Comunicación Social maguisen@gmail.com . Síntesis del Proyecto Especial de Innovación Social (PEIS) 2023 El CONICET y la Fundación Síndrome de Rett (SinRett) Argentina se han unido para desarrollar juntos el proyecto "Punto tecnológico para la accesibilidad de personas con síndrome de Rett y otras condiciones neurológicas", ganador de la convocatoria Proyectos Especiales de Innovación Social (PEIS) 2023 organizada por la Gerencia de Vinculación Tecnológica del Ministerio de Ciencia Tecnología e Innovación de la Nación (MinCyT).
Las personas con síndrome de Rett y otras condiciones neurológicas, en particular personas con dificultades motrices y en el habla, encuentran limitaciones en el acceso a la comunicación y, por lo tanto, a la educación. El objetivo de este proyecto es crear un punto tecnológico para el desarrollo de estrategias de accesibilidad comunicacional y educativa destinado a este grupo poblacional y haciendo uso del potencial de las Tecnologías de la Información y la Comunicación (TIC), entre ellas los sistemas de tracking eye, aplicadas a la comunicación aumentativa.
Partiendo de un grupo 95 personas, durante el año 2024 se trabajará con una selección de casos de acuerdo a las condiciones de los mismos y la factibilidad de desarrollo de las estrategias. Cada una de ellas abarcará las siguientes fases: a. evaluación de accesibilidad, b. compilación funcional de tecnologías, c. capacitación de recursos humanos del territorio, y d. seguimiento y actualización. Como resultado se busca el incremento de la accesibilidad del grupo destinatario, la federalización del impacto social del proyecto, el desarrollo científico tecnológico, y la implementación, transferencia y generación de conocimiento.
Adoptaremos una perspectiva federal, situada e interdisciplinaria, y una modalidad mixta: presencial (con sede física en “EquipoNeuro” Rosario, Santa Fe); itinerante (movilizándonos dentro de Rosario, sus cercanías y a localidades más alejadas dentro del país); remota (on line); e itinerante (en red con profesionales competentes en accesibilidad cercanos a la localidad donde reside la persona destinataria de la estrategia). El carácter del proyecto es esencialmente asociativo. Ante el desafío propuesto, se une al CONICET y SinRett Argentina el Centro de rehabilitación, estimulación temprana y servicio de apoyo a la integración escolar “EquipoNeuro” ubicado en Rosario, Santa Fe; y las siguientes instituciones del Sistema Nacional de Ciencia, Tecnología e Innovación (SNCTI): el Centro de Investigación y desarrollo en Tecnologías Especiales (CeDITE) de la Universidad Tecnológica Nacional (UTN) Facultad Regional Rosario (FRRo), la cátedra de “Patología y terapéutica fonoaudiológica en la parálisis cerebral” de la Escuela de Fonoaudiología de la Facultad de Medicina de la Universidad Nacional de Rosario (UNR), y el Programa Educación y Sociedad. Hacia una mayor inclusión educativa, de la Secretaría de Extensión y Cultura de la Universidad Nacional del Litoral con sede en la Facultad de Humanidades y Ciencias (UNL). El equipo de trabajo se compone actualmente por 20 personas, entre ellas: tecnólogos/as, terapeutas, investigadores/as especialistas en accesibilidad, educadores/as, y familiares del grupo objetivo; y se caracteriza por ser interdisciplinario, paritario en cuanto al género y diverso. La sinergia de saberes tiene el potencial necesario para generar una dinámica innovadora hacia la cumplimentación del objetivo propuesto. Comprometidos/as con el ejercicio de derechos de las personas con discapacidad, avanzamos en la implementación de este proyecto dispuestos a incluir en la red que lo sostiene a quienes compartan nuestros principios y estén dispuestos a trabajar para su fortalecimiento y expansión.
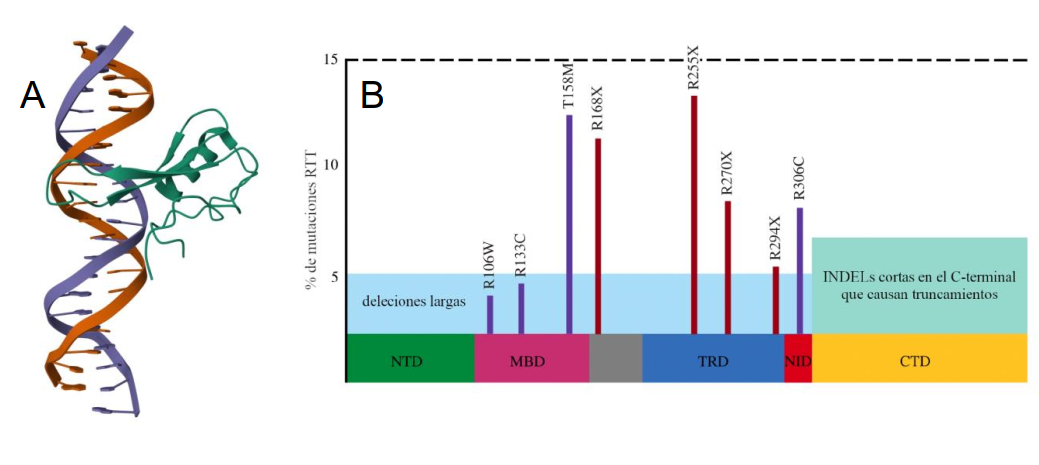
Figura 6: La proteína MeCP2 su función y sus variantes. Estructura del dominio de unión a metil-CpG de MeCP2 humana en complejo con una secuencia de ADN metilada (PDB ID: 3C2I). (B) Mutaciones más frecuentes de MeCP2 en pacientes con síndrome de Rett, un esquema de MeCP2 con sus dominios funcionales (tomado de la Tesis de Licenciatura de la Lic. M. Belén Cardillo).
Discinesia Ciliar Primaria (DCP, ORPHA:244)
Asociación en Argentina: Grupo de pacientes DCP
Contacto: argentina.dcp@gmail.com
Título del Proyecto: Análisis de la resistencia a antibióticos y virulencia en Staphylococcus aureus, Moraxella catarrhalis y Pseudomonas aeruginosa en infecciones polimicrobianas en pacientes con diagnóstico de discinesia ciliar primaria: diferencias y similitudes con la fibrosis quística.
Etapa en la que se encuentra el proyecto: iniciado
Necesidad de financiamiento: Si
Modalidad: Análisis de aislamientos bacterianos provenientes de pacientes con DCP y comparación con aquellos derivados de FQ. Integración con características clínicas.
Integrantes: Paula M. Tribelli1,2, Juan Ballinoti3; Lorena Ibarra3, Pablo M. Cassanelli4, Rosana Pereda4, Silvina Smith4, Stefania Robaldi1, Maria Constanza Paustasso1, Mateo Tripoloni2 y Georgina Davis5
Filiaciones: 1IQUIBICEN-CONICET; 2Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires, 3Hospital General de Niños Dr. Ricardo Gutierrez, 4Hospital General de Niños Dr. Pedro Elizalde, 5ALAPA Argentina.
La discinesia ciliar primaria (DCP) es una enfermedad genética poco frecuente que afecta la función de los cilios, estructuras celulares presentes revestimiento de las vías respiratorias, los oídos y los senos paranasales [29]. La función principal de estas estructuras es la movilización de partículas que pueden ingresar desde el exterior incluyendo a los microorganismos y de la mucosidad provocada por las distintas células [30]. La disfunción de estas estructuras provoca una depuración mucociliar defectuosa y por ende un microambiente propicio para el desarrollo de infecciones recurrentes del tracto respiratorio superior e inferior, así como en oídos [31]. Los pacientes con diagnóstico de esta enfermedad sufren infecciones persistentes o recurrentes que incluyen tanto especies Gram positivas como Staphylococcus aureus y Gram negativas como Pseudomonas aeruginosa y Moraxella catarrhalis [31]. Sin embargo, estas infecciones suelen ser consideradas polimicrobianas ya que más de un microorganismo puede estar coexistiendo o formando comunidades multi-especies con relaciones o interacciones neutrales, mutualistas o antagonistas [32].
En este contexto las interacciones entre microorganismos durante procesos infecciosos influyen en la virulencia, así como en la formación de biofilms, tolerancia y resistencia a antibióticos, la progresión clínica y el resultado final (Figura 7) [32, 33]. Es por esto por lo que se necesitan estudios que analicen las características de los microorganismos dominantes o más abundantes, pero tomando en cuenta los microorganismos presentes que puedan influir en sus características fisiológicas, de virulencia y principalmente de resistencia a antibióticos. En el curso de las infecciones polimicrobianas la virulencia y la resistencia a antibióticos se modifica en comparación a las infecciones monoespecie, favoreciendo la persistencia de alguna/s de la/s especie/s que las componen. La presencia de M. catharralis en la DCP provoca diferencias en las infecciones polimicrobianas respecto a la fibrosis quística. El objetivo general del proyecto es evaluar si los aislamientos pertenecientes a pacientes coinfectados poseen resistencia a antibióticos, virulencia y capacidad de competencia diferentes a los aislamientos provenientes de pacientes monoinfectados. Realizamos una batería de experimentos in vitro para entender los fenotipos de las cepas bacterianas circulantes en nuestro país y los mecanismos fisiológicos y moleculares que llevan a la modificación en la virulencia y resistencia a antibióticos cuando más de un microorganismo se encuentra causando la infección.
El objetivo final de nuestra investigación es obtener información que pueda ser utilizada traslacionalmente para el manejo de infecciones polimicrobianas en pacientes con DCP. Creemos que es relevante ya que gran parte de la información que se posee hasta el momento es derivada del tratamiento de los pacientes con fibrosis quística (FQ). Sin embargo, la patofisiología de la FQ y la DCP no son idénticas por lo que no necesariamente las infecciones van a presentar la misma dinámica. Nos encontramos recolectando aislamientos bacterianos de las tres especies más frecuentemente aisladas provenientes de pacientes pediátricos que se atienden en el Hospital Gutierrez (DCP; PRIISA BA: 10506) y el Hospital Pedro Elizalde (FQ; PRIISA BA: 4466) y analizando la virulencia y la resistencia a antibióticos utilizados en la práctica médica. Es importante destacar que las infecciones crónicas son la mayor causa de morbilidad en los pacientes con DCP y de morbi-mortalidad en los pacientes con FQ.

Figura 7: Nuestro proyecto se centra en el estudio de las infecciones polimicrobianas en la discinesia ciliar primaria (DCP), en particular nos enfocamos en las interacciones entre Pseudomonas aeruginosa y Staphylococcus aureus teniendo en cuenta la presencia o no de Moraxcella catarrhalis y cómo afecta a la virulencia y la resistencia a antibióticos. Nos interesa entender las diferencias de las infecciones en DCP en relación con la más estudiada Fibrosis Quística, en donde la presencia de M. catarrhalis es menos frecuente.
Referencias:
1. Taruscio D and WA Gahl, Rare diseases: challenges and opportunities for research and public health. Nat Rev Dis Primers, 2024. 10(1): p. 13.
2. Villalon-Garcia I, et al., Precision Medicine in Rare Diseases. Diseases, 2020. 8(4).
3. Giugliani R, et al., Opportunities and challenges for newborn screening and early diagnosis of rare diseases in Latin America. Front Genet, 2022. 13: p. 1053559.
4. Hua Y, et al., Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet, 2008. 82(4): p. 834-48.
5. Hua Y, et al., Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev, 2010. 24(15): p. 1634-44.
6. Hua Y, et al., Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature, 2011. 478(7367): p. 123-6.
7. Schor IE, et al., Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci U S A, 2009.106(11): p. 4325-30.
8. Schor IE, et al., Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. EMBO J, 2013. 32(16): p. 2264-74.
9. Allo M, et al., Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol, 2009. 16(7): p. 717-24.
10. Allo M, et al., Argonaute-1 binds transcriptional enhancers and controls constitutive and alternative splicing in human cells. Proc Natl Acad Sci U S A, 2014. 111(44): p.15622-9.
11. Dujardin G, et al., How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell, 2014. 54(4): p. 683-90.
12. Garcia-de-Teresa B, A Rodriguez, and S Frias, Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes (Basel), 2020. 11(12).
13. Hodson C, et al., Structure of the human FANCL RING-Ube2T complex reveals determinants of cognate E3-E2 selection. Structure, 2014. 22(2): p. 337-44.
14. Rennie ML, et al., Differential functions of FANCI and FANCD2 ubiquitination stabilize ID2 complex on DNA. EMBO Rep, 2020. 21(7): p. e50133.
15. Kowal P, et al., Structural determinants of human FANCF protein that function in the assembly of a DNA damage signaling complex. J Biol Chem, 2007. 282(3): p. 2047-55.
16. Wang S, et al., Structure of the FA core ubiquitin ligase closing the ID clamp on DNA. Nat Struct Mol Biol, 2021. 28(3): p. 300-309.
17. Tryon R, et al., Phenotypic variability in patients with Fanconi anemia and biallelic FANCF mutations. Am J Med Genet A, 2017. 173(1): p. 260-263.
18. Zareifar S, et al., A novel frame-shift deletion in FANCF gene causing autosomal recessive Fanconi anemia: a case report. BMC Med Genet, 2019. 20(1): p. 122.
19. Lill R, From the discovery to molecular understanding of cellular iron-sulfur protein biogenesis. Biol Chem, 2020. 401(6-7): p. 855-876.
20. Fox NG, et al., Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat Commun, 2019. 10(1): p.2210.
21. Pandolfo M, Molecular genetics and pathogenesis of Friedreich ataxia. Neuromuscul Disord, 1998. 8(6): p. 409-15.
22. Frecot DI, T Froehlich, and U Rothbauer, 30 years of nanobodies - an ongoing success story of small binders in biological research. J Cell Sci, 2023. 136(21).
23. Parrasia S, et al., Peptides as Pharmacological Carriers to the Brain: Promises, Shortcomings and Challenges. Mol Pharm, 2022. 19(11): p. 3700-3729.
24. Sheikh SR, et al., The role of brainstem biopsy and targeted therapies in pediatric diffuse midline glioma/diffuse intrinsic pontine glioma. Front Oncol, 2024. 14: p.1504440.
25. Kamran N, et al., Current state and future prospects of immunotherapy for glioma. Immunotherapy, 2018. 10(4): p. 317-339.
26. Van den Veyver IB and HY Zoghbi, Genetic basis of Rett syndrome. Ment Retard Dev Disabil Res Rev, 2002. 8(2): p. 82-6.
27. Bajikar SS, et al., Acute MeCP2 loss in adult mice reveals transcriptional and chromatin changes that precede neurological dysfunction and inform pathogenesis. Neuron, 2025. 113(3): p. 380-395 e8.
28. Lombardi LM, SA Baker, and HY Zoghbi, MECP2 disorders: from the clinic to mice and back. J Clin Invest, 2015. 125(8): p. 2914-23.
29. Baz-Redon N, et al., Implementation of a Gene Panel for Genetic Diagnosis of Primary Ciliary Dyskinesia. Arch Bronconeumol (Engl Ed), 2021. 57(3): p. 186-194.
30. Horani A and TW Ferkol, Understanding Primary Ciliary Dyskinesia and Other Ciliopathies. J Pediatr, 2021. 230: p. 15-22 e1.
31. Wijers CD, JF Chmiel, and BM Gaston, Bacterial infections in patients with primary ciliary dyskinesia: Comparison with cystic fibrosis. Chron Respir Dis, 2017. 14(4): p. 392-406.
32. Nair N, et al., Impact of Staphylococcus aureus on pathogenesis in polymicrobial infections. Infect Immun, 2014. 82(6): p. 2162-9.
33. Michelsen CF, et al., Staphylococcus aureus alters growth activity, autolysis, and antibiotic tolerance in a human host-adapted Pseudomonas aeruginosa lineage. J Bacteriol, 2014. 196(22): p. 3903-11.
ISSN 1666-7948 www.quimicaviva.qb.fcen.uba.ar |
Revista QuímicaViva Volumen 24, Número 1, Abril de 2025 |
Publicado en:
Vol 24, Nro 1
Abril de 2025
Identificador: E0292
DOI:10.62167/qv.e0292
Tipo: Sección especial
Recibido en: 24/02/2025
Aceptado en: 21/04/2025
Contacto: ALAPA