Reevaluación de los residuos cisteína en el señalamiento redox
Martin Aran, Santiago Mora Garcia, Laura Rimmaudo y
Ricardo A. Wolosiuk
Instituto Leloir, Depto. Quìmica Biológica-FCEN-UBA, IIBBA-CONICET,
Patricias Argentinas 435, C1405 Buenos Aires, Argentina
e-mail: <rwolosiuk@leloir.org.ar>
Recibido el 02/10/2009. Aceptado el 14/12/2009.
Resumen
La formación, escisión e isomerización de los puentes
disulfuro juega un rol importante en la estructura y la función de las
proteínas. Sin embargo, las especies reactivas de oxígeno pueden aumentar el
estado de oxidación del átomo de azufre en las cisteínas generando los
oxiácidos sulfénico (−R−SOH), sulfínico (−R−SO2H) y sulfónico (−R−SO3H). Las
peroxirredoxinas, ubicua familia de peroxidasas desprovistas del grupo
prostético hemo, utilizan esta modificación post-traduccional de las
cisteínas para catalizar la reducción del peróxido de hidrógeno o iniciar la
respuesta al estrés oxidativo [Prx−Cys−SH + H2O2 → Prx−Cys−SOH + H2O]. La
restitución del grupo tiol a las peroxirredoxinas procede en dos etapas
adicionales (resolución, reducción) que requieren la participación de
sistemas complejos para la transferir el poder reductor del NADPH. Además,
una nueva proteína, la sulfirredoxina, cataliza la reducción del grupo
sulfínico a sulfénico en un proceso dependiente de ATP. En este contexto, la
modificación de las interacciones no-covalentes y la transformación de las
uniones covalentes, e.g. fosforilación, acetilación, asisten a las
peroxirredoxinas en la regulación de los procesos biológicos. Sobre estas
bases, las actividades de las peroxirredoxinas están reguladas no sólo por
los múltiples estados de oxidación del átomo de azufre sino también por la
integración de la química no-redox. Esta característica dota a las
peroxirredoxinas de mecanismos versátiles para percibir los múltiples
cambios celulares y generar diferentes especies moleculares como respuesta
adecuada a los estímulos ambientales.
Palabras clave: especies reactivas de oxígeno / estrés oxidativo / sulfénico
/ sulfínico / sulfónico / intercambio tiol-disulfuro / glutationilación de
proteínas / peroxirredoxina / fosforilación / acetilación /
_____________________________________________________
Summary
The formation, cleavage and isomerization of disulfide bonds play an
important role in protein structure and function. However, reactive oxygen
species may increase the oxidation state of the sulfur atom in cysteines
yielding the oxyacids sulfenic (−R−SOH), sulfinic (−R−SO2H) and sulfonic
(−R−SO3H). Peroxiredoxins, the
ubiqutous family of peroxidases devoid of the heme prosthetic group, utilize
this post-translational modification of cysteines for catalyzing the
reduction of hydrogen peroxide or triggering the response to the oxidative
stress [Prx−Cys−SH + H2O2 → Prx−Cys−SOH + H2O]. The restitution of the thiol
group to peroxiredoxins proceeds in two steps (resolution, reduction) that
require the participation of complex systems for transferring the NADPH
reductant power. Moreover, a novel protein, sulfiredoxin, catalyzes the
ATP-mediated reduction of the sulfinic group to sulfenic. In this context,
the modification of non-covalent interactions and the transformation of
covalent bonds, e.g. phosphorylation, acetylation, assist peroxiredoxins in
the regulation of biological process. On this basis, the activities of
peroxiredoxins are regulated through not only the oxidation states of the
sulfur atom but also the non-redox chemistry. This feature endows
peroxiredoxins with versatile mechanisms to perceive multiple cellular
changes and generate different molecular species as adecuate response to
environmental stimuli.
Keywords: reactive oxygen species / oxidative stress / sulfenic / sulfinic /
sulfonic / tiol-disulfide exchange / protein glutathionylation /
peroxiredoxin / phosphorylation / acetylation /
Las células utilizan las reacciones de óxido-reducción (redox) para promover
y modular el crecimiento de los organismos. Procesos biológicos tan
diferentes como la inflamación y la fotosíntesis requieren inevitablemente
la transferencia de electrones o de hidrógenos ([H] ≡ H+ + e−) para mantener
las proporciones adecuadas de las formas [oxidada]/[reducida] de una especie
química. Esta característica sugiere que las reacciones redox sirven para
detoxificar eficientemente las especies reactivas o cumplir un rol en la
regulación de los caminos metabólicos. La consecuencia inmediata de estas
posibilidades es que la disfunción de los procesos redox estará asociada con
determinadas patologías.
La señalización redox exhibe diferencias notables con los mecanismos que
utilizan la química no-redox en la regulación celular. Un estímulo provoca
solamente una respuesta “on/off” cuando los aminoácidos de las proteínas
exhiben una única modificación post-traduccional (e.g. fosforilación,
acetilación). En consecuencia, la respuesta paulatina a los estímulos
requiere la utilización de varios sitios regulatorios en una proteína y/o la
activación simultánea de varios pasos metabólicos. De manera que la
respuesta gradual y continua en un camino de señalización basado en
mecanismos “on/off” depende de un complejo sistema de proteínas y/o residuos
y no de un residuo proteico en particular.
La presencia de una atmósfera rica en O2 y el uso de esta molécula como
oxidante final de las cadenas respiratorias por una parte significativa de
la biósfera implica un flujo continuo de electrones hacia el O2 para formar
H2O mediante oxidasas específicas. Este proceso está inevitablemente
acompañado por la formación de especies derivadas de la reducción
monoelectrónica, conocidas colectivamente como especies reactivas de oxígeno
(ROS: reactive oxygen species). Si bien las ROS afectan de forma general
sitios reactivos en los hidratos de carbono, los lípidos y los ácidos
nucleicos, durante la última década numerosos estudios revelaron que las ROS
también modulan el estado de oxidación del azufre en residuos cisteína
específicos de las proteínas. La notable capacidad del azufre para adoptar
múltiples estados de oxidación genera no sólo los oxiácidos sulfénico
(RSOH), sulfínico (RSO2H), y sulfónico (RSO3H) sino también otras especies
oxidadas [e.g. tiosulfinatos (disulfuro-S-monóxidos, RS(O)SR´)], revelando
que la función de los tioles en las cisteínas no está circunscripta
exclusivamente a la formación y ruptura de puentes disulfuro. La
flexibilidad de un único residuo cisteína para adoptar diferentes
modificaciones post-traduccionales sugiere que
varias de estas especies pueden participar en el señalamiento redox
intracelular (1). Sobre estas bases, el reconocimiento del estímulo y su
transducción no residen en un sistema complejo ni en múltiples
modificaciones de una proteína sino en un único residuo de aminoácido.
El objetivo de esta revisión es describir los mecanismos redox que captan
estímulos ambientales para generar las señales que llevan a la resistencia o
la tolerancia celular al estrés impuesto. La primera sección resume las
diferentes ROS que son generadas intracelularmente y que inician la
transducción de la señal. La segunda sección contiene un panorama de los
cambios en los estados de oxidación del átomo de azufre de las cisteínas que
provocan cambios en las estructuras secundaria, terciaria y cuaternaria de
las proteínas. Evidentemente, las proteínas poseen otros residuos, tales
como metionina e histidina, susceptibles a la modificación mediante química
redox (2,3), pero en esta revisión destacaremos la importancia de los
residuos cisteína por su extrema sensibilidad a los procesos redox y su
vinculación a ciertas patologías. La tercera sección describe brevemente los
sistemas enzimáticos que protegen a las células de los efectos letales
causados por la formación excesiva de ROS. En este contexto, el énfasis fue
puesto sobre las características bioquímicas de una familia de proteínas
antioxidantes, las peroxirredoxinas (Prx), porque los estudios de la última
década revelaron su importancia no sólo en la defensa antioxidante sino
también en la señalización redox.
Especies reactivas de oxígeno
Hace 3.5 x 109 años, las cianobacterias comenzaron a utilizar la energía
solar para incorporar el carbono inorgánico ambiental [CO2, HCO3−] a las
cadenas alifáticas de los azúcares [−CH2−HCOH−] empleando el H2O como
reductor [H2O → 4 H+ + O2 + 4 e−]. Este evento llevó a la acumulación de O2
en la atmósfera (actualmente ca. 21%) y a la aparición de los sistemas
respiratorios que dependen del O2. Sin embargo, la molécula de O2 es
moderadamente reactiva por la presencia de dos electrones desapareados en el
orbital exterior (Figura 1A).
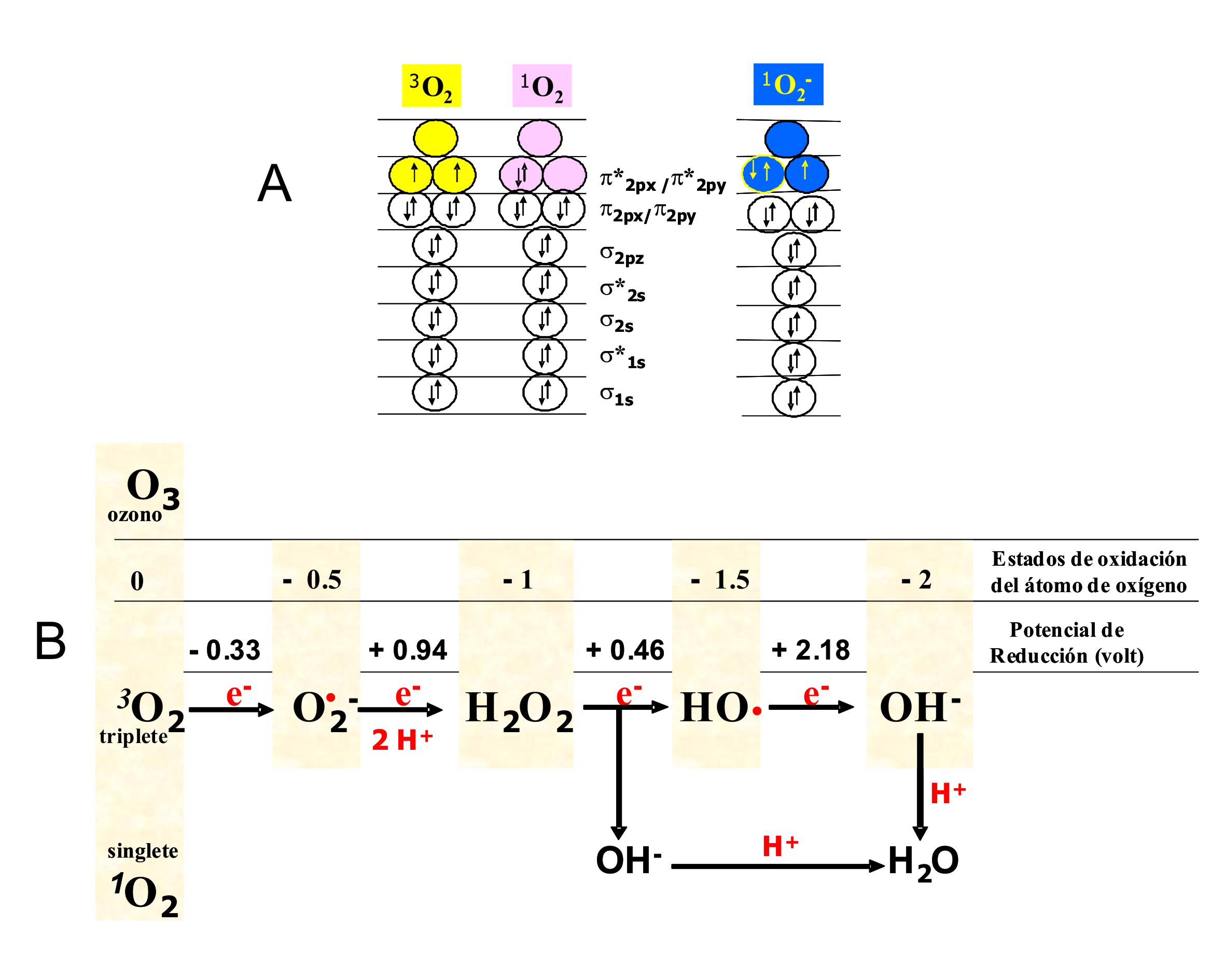
Figura 1. (A) Orbitales moleculares del O2. (B) Reducción monoelectrónica del oxígeno. Estados de oxidación y potenciales redox.
La reducción parcial del O2 atmosférico [oxígeno triplete (3O2)] produce dos
categorías de ROS: los radicales, átomos o grupo de átomos que tienen uno o
más electrones desapareados [anión superóxido (O2•−), radical hidroxilo
(HO•)], y las especies neutras [ozono (O3), oxígeno singlete (1O2), peróxido
de hidrógeno (HO−OH), hidroperóxidos orgánicos (RO−OH)]. Un amplio rango de
macromoléculas (DNA, lípidos, proteínas) es modificado cuando la síntesis de
las ROS no es controlada apropiadamente. El radical hidroxilo tiene una
existencia extremadamente corta mientras que los hidroperóxidos son
relativamente estables (4). Esta característica indica que el primero
reaccionaría con las dianas en el sitio donde es generado mientras que los
segundos podrían difundir para actuar sobre otros compartimentos o células.
Generación de especies reactivas de oxígeno
Las células de los diferentes organismos que pueblan la bioesfera reciben
numerosos estímulos provocados por su interacción con los seres vivos
(estrés biótico) o los eventos físico-químicos (estrés abiótico). Una de las
respuestas más extendidas ante estos factores exógenos es un aumento de los
niveles intracelulares de ROS. Por ello, las células han desarrollado una
serie de mecanismos enzimáticos complejos que generan y consumen las ROS
para mantener sus concentraciones intracelulares bajo control.
Es ampliamente conocido que la respiración aeróbica en las membranas
mitocondriales reduce parcialmente el O2 generando el anión superóxido O2•−.
Cerca del 2% del O2 consumido en la respiración mitocondrial es destinado a
la producción del O2•− (5). Por otra parte, la exposición de un sinnúmero de
células y tejidos a una variedad de agentes físicos, químicos y biológicos
produce O2•− en una reacción catalizada por la NADPH oxidasa asociada a la
membrana plasmática [NADPH + 2 O2 → NADP+ + 2 O2•− + H+] (Figura 2).
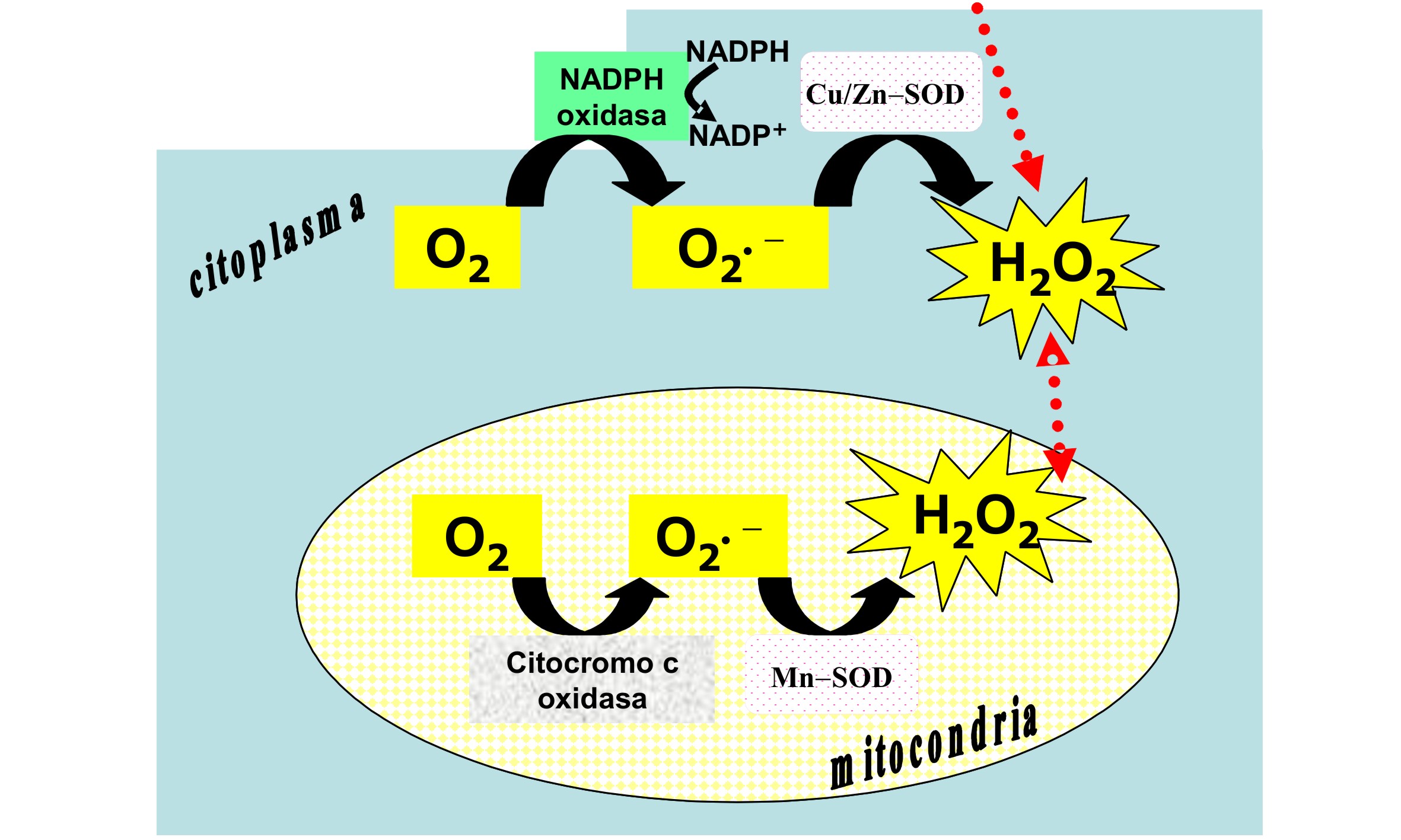
Figura 2. Formación intracelular y extracelular de H2O2 en el
sistema inmune de mamíferos.
Por otro lado, numerosos efectos citotóxicos tienen su origen
en la extrema capacidad oxidante del H2O2 [H2O2 + 2 H+ + 2 e− → 2 H2O; Eo =
+ 1.76 volt]. La síntesis del H2O2 aumenta como réplica a numerosos estreses
regulando procesos biológicos tan diversos como la respuesta inmune en los
mamíferos y el cierre de los estomas en las plantas. Es evidente que la
utilización de un compuesto citotóxico como molécula señal implica (i) un
ajustado control de su síntesis y degradación y (ii) su adecuada
localización y nivel celular. Las enzimas antioxidantes actúan como sensores
de los niveles de peróxidos intracelulares y, en consecuencia, modulan una
pléyade de procesos biológicos que responden al H2O2.
Numerosas evidencias indican que la ruptura del anión superóxido [2 O2•− + 2
H+ → H2O2 + O2], espontánea o mediada por la superóxido dismutasa, implica
una mayor producción de H2O2. Varios factores de crecimiento y citoquinas
(PDGF, EGF, insulina, angiotensina II, TNFα) estimulan la activación de las
NADPH oxidasas causando un aumento en el nivel celular de H2O2. Sin embargo,
la generación celular de H2O2 no procede exclusivamente por esta vía
metabólica porque los niveles de H2O2 también aumentan mediante mecanismos
independientes de la NADPH oxidasa. Por ejemplo, la producción de
extracelular de H2O2 aumenta cuando ligandos específicos interaccionan con
los receptores de hematopoyetina pero disminuye cuando uno de los receptores
ha sido mutado (6). Este H2O2 difunde a través de la membrana plasmática
para participar en la modulación del señalamiento redox (Figura 2). En
conjunto estos estudios revelan que la producción de H2O2 tiene lugar por
mecanismos que implican a la cadena respiratoria, la NADPH oxidasa y las
interacciones receptor-ligando.
En un contexto biológico totalmente diferente, los cloroplastos en las hojas
de las plantas superiores poseen caminos metabólicos particulares para la
producción de ROS. Bajo iluminación, el oxígeno singlete 1O2 es producido
por la clorofila triplete en los centros de reacción del fotosistema II y en
el sistema antena (7).
1Chl →(LUZ)→ 3Chl
3Chl + 3O2 → Chl + 1O2
Utilizando el 1O2, la cadena fotosintética de transporte de electrones tiene
la capacidad para generar el O2•− y el H2O2. Sin embargo, las células
foliares poseen además una via metabólica particular, denominada
fotorrespiración, para producir cantidades apreciables de H2O2 (Figura 3).
En los cloroplastos, la actividad oxigenasa de la ribulosa-1,5-bisfosfato
carboxilasa-oxigenasa (Rubisco) genera 2-fosfoglicolato que es transformado
a glicolato. Este úlimo compuesto fluye de los cloroplastos hacia los
peroxisomas, donde es oxidado por la flavoenzima glicolato oxidasa con la
producción simultánea de H2O2.
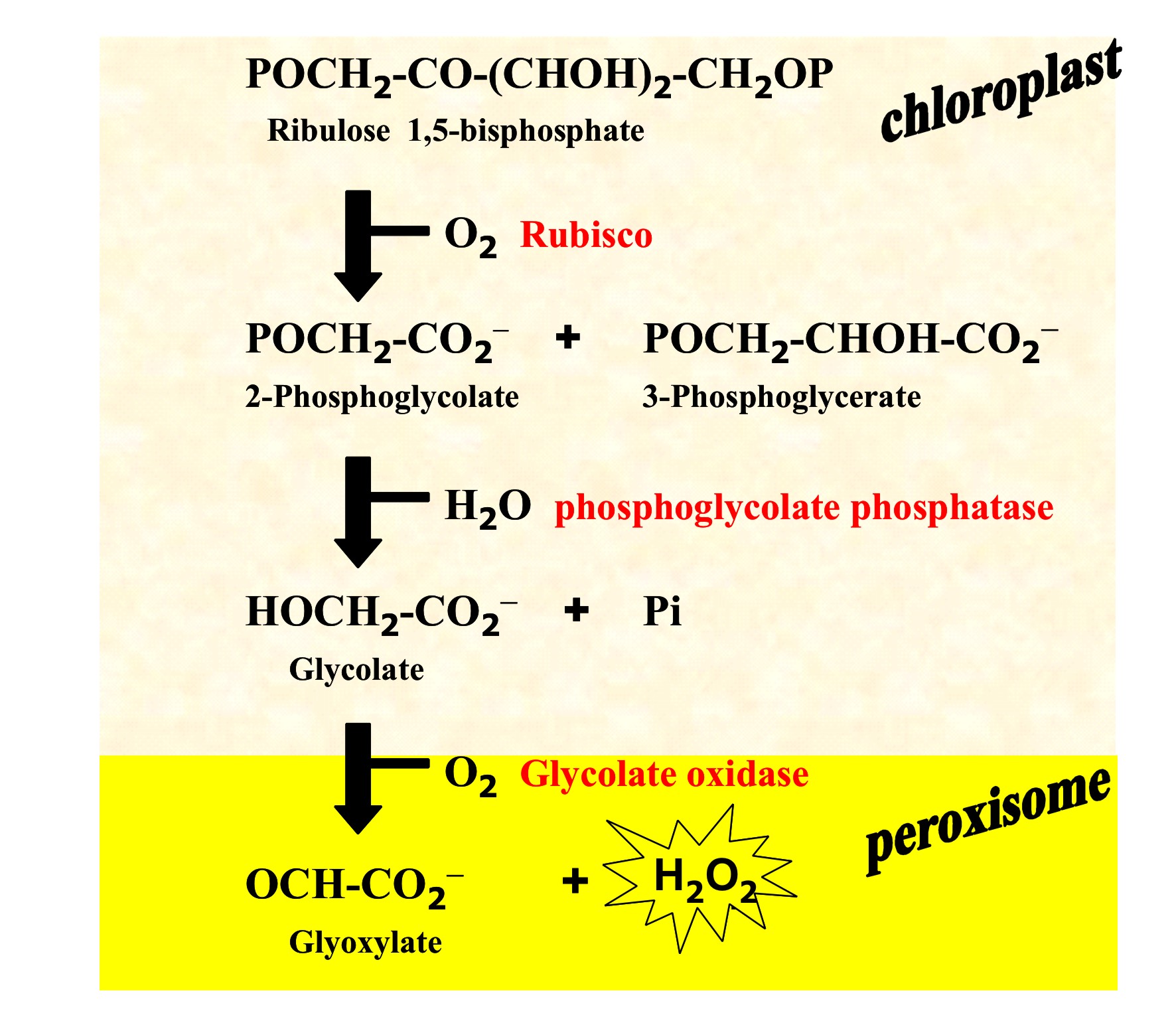
Figura 3. Procesos iniciales de la fotorrespiración en las hojas de las
plantas superiores.
Las células animales y vegetales generan H2O2 como parte de
los mecanismos de tolerancia y defensa frente al desafío provocado por
agentes físicos o patógenos. Los últimos a su vez replican utilizando el
H2O2 para incrementar los niveles de los factores de transcripción que
regulan la expresión de genes implicados en la detoxificación. Los estudios
de señalamiento redox han alcanzado un notable desarrollo, impulsados por la
idea de que los procesos oxidativos pueden ser utilizados para regular
múltiples respuestas. En este aspecto, el H2O2, ampliamente conocido por sus
acciones citotóxicas, ahora constituye un modulador importante en la
transducción de señales en los diferentes compartimentos de los eucariotes.
Modificación de las cisteínas en las cadenas polipeptídicas
Las alteraciones causadas por las ROS sobre el DNA, los lípidos y las
proteínas son frecuentes en los organismos aeróbicos, tanto procarióticos
como eucarióticos, siendo responsables de las patologías relacionadas al
envejecimiento de los animales y a la senescencia de las plantas. Aunque una
vasta literatura describe las transformaciones mediadas por las ROS en las
moléculas de DNA y los lípidos en los sistemas biológicos, la revisión
actual está circunscripta a los efectos ocasionados por las transformaciones
redox sobre los aminoácidos de las proteínas. Estas modificaciones post-traduccionales
constituyen actualmente los mecanismos más importantes para vincular las ROS
con los caminos de transducción de señales porque diferentes estados de
oxidación de residuos específicos determinan las funciones de las proteínas
y sus uniones a los ligandos. Las proteínas implicadas en la señalización
deben cumplir dos requisitos para provocar una
respuesta eficiente: los residuos regulatorios deben residir en posiciones
específicas y las transformaciones del estado de oxidación deben ser rápidas
y reversibles.
Mientras las modificaciones post-traduccionales clásicas (e.g. fosforilación,
acetilación) provocan un único cambio en el residuo afectado, los diferentes
estados de oxidación del átomo de azufre en los residuos cisteína y
metionina permiten generar varias formas de una proteína. Esta multiplicidad
de especies sobre un mismo residuo permite sugerir la existencia de puntos
de ramificación cuya efectividad en la respuesta a los estímulos dependerá
de las características termodinámicas y cinéticas. La regulación
termodinámica asume que todos los pares tiol-disulfuro celulares están en
equilibrio de manera que la proporción [−Cys-SH(oxidado)]/[−Cys-SH(reducido)]
en una proteína estará determinada por el potencial redox celular (Figura 4,
via a). Cuando el buffer redox celular altera su proporción
[oxidado]/[reducido], varía el contenido de puentes disulfuro en las
proteínas reguladoras y éstas inician la transmisión de la señal que provoca
la respuesta celular. En esta función, dos características celulares son
importantes para el establecimiento del buffer redox celular. Por un lado la
composición, ya que el par glutation oxidado/glutation reducido (GSH, γ-glutamyl-cysteinyl-glycine
[2 GSH + 2H+ + 2e− → GSSG; Eo=−240 mV]) y el par dehidroascorbato/ascorbato
constituyen los principales buffer redox en las células animales y
vegetales, respectivamente. Por otro lado la localización intracelular y el
estado de desarrollo, por cuanto el potencial redox del par GSH/GSSG es –
150 mV y – 300 mV en el retículo endoplásmico y las mitocondrias,
respectivamente mientras que en el citoplasma de células proliferativas y
apoptóticas es de – 260 mV y – 170 mV, respectivamente.
Sin embargo, el intercambio tiol-disulfuro entre estas moléculas es lento y
aparentemente las células no mantienen un rápido equilibrio
termodinámico.Por esta razón, enzimas específicas [e.g. tiorredoxina (Trx),
glutarredoxina, protein-disulfuro isomerasa] catalizan el intercambio
tiol-disulfuro para mantener la homeostasis celular cuando un estímulo
altera al buffer redox.
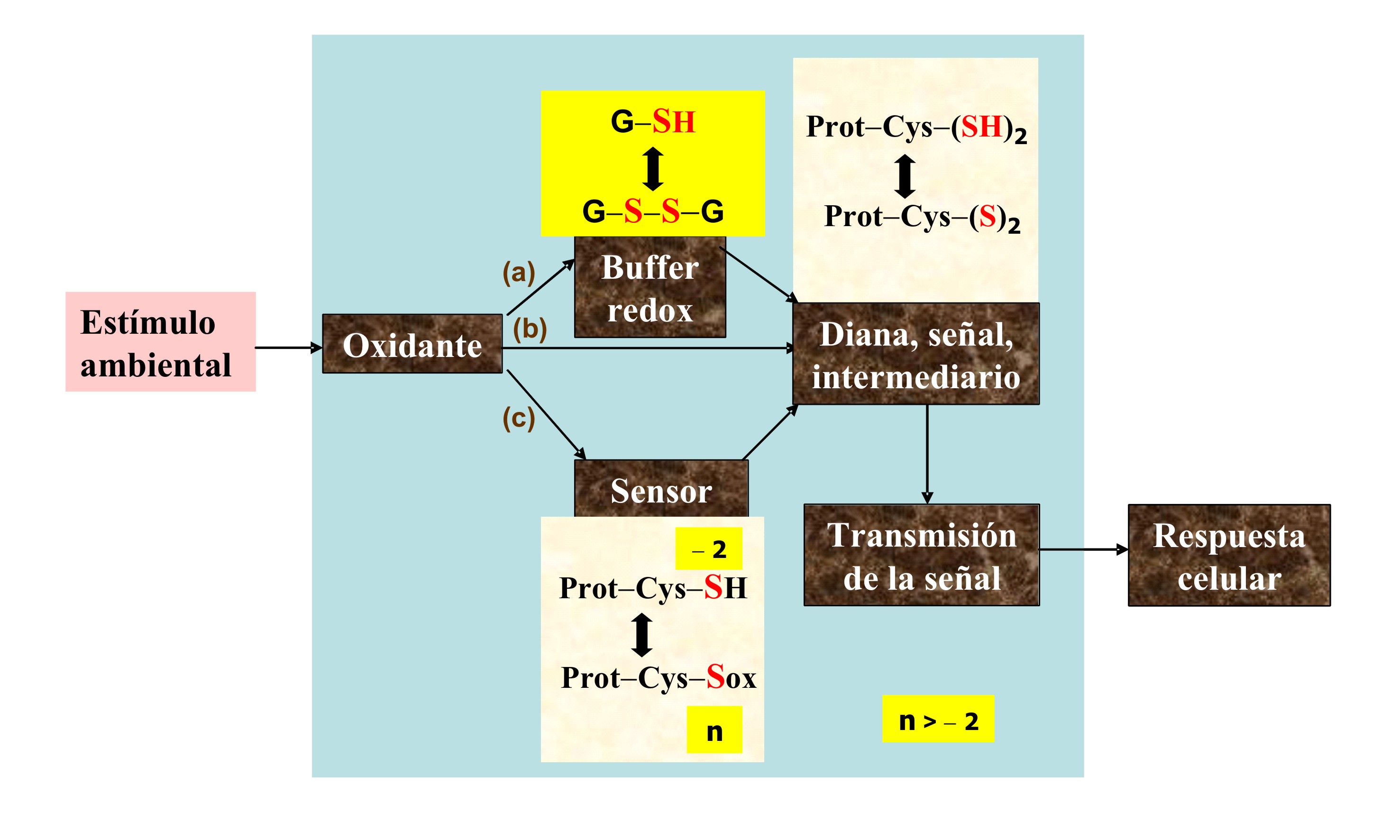
Figura 4. Mecanismos para el señalamiento redox.
Por otra parte, las propiedades cinéticas de las dianas pueden facilitar
respuestas específicas a las ROS. La oxidación transitoria de estas dianas
las capacita para desencadenar una respuesta y luego retornar al estado
basal mediante la reducción enzimática (Figura 4, via b). De esta forma
actúa un grupo de proteínas denominadas sensores, cuyos tioles son
extremadamente sensibles a los oxidantes. Producida la oxidación, el sensor
afecta la funcionalidad de otras dianas mediante interacciones no-covalentes
proteína-proteína o la transformación covalente de las dianas mediante el
intercambio tiol-disulfuro (Figura 4, via c).
Intercambio tiol-disulfuro
A pesar de las nuevas evidencias sobre las múltiples variaciones en una
única cisteína, el intercambio tiol-disulfuro [R1−SH + R2−S−S−R3 ⇔ R1−S−S−R2
+ R3−SH] es aún el mecanismo más estudiado. La formación de los puentes
disulfuro no sólo estabiliza las proteínas extracelulares, protege contra la
inactivación, determina la asociación con otras proteínas sino también
regula las funciones de las proteínas. Un variado grupo de enzimas
denominadas colectivamente protein-disulfuro óxido-reductasas (PDOR)
cataliza el intercambio tiol-disulfuro. La disponibilidad de las secuencias
genómicas completas y los estudios de proteómica han revelado dos rasgos
relevantes en esta ubicua familia de proteínas. Primero, la presencia del
motivo -CXXC- en su centro activo, cuyas cisteínas participan en un proceso
redox cíclico que forma un puente disulfuro intracatenario con diferentes
tendencias catalíticas: las Trx y las SHS−S−SHSox(SH)(S)SH(S−S−(SHSoxSHSox
glutarredoxinas derivan hidrógenos (2 H+ + 2 e−) hacia la ruptura de
los puentes disulfuro (E. coli Trx, Eo = − 270 mV; E. coli Grx, Eo = − 233
mV) mientras que las DsbA bacterianas y protein-disulfuro isomerasas
eucarióticas remueven hidrógenos de los tioles de las proteínas dianas (E.
coli DsbA, Eo = − 106 mV). Segundo, todos los miembros de esta superfamilia
comparten una estructura terciaria común compuesta de cuatro cintas-β
rodeadas de tres α-hélices. Esta unidad básica, denominada plegamiento Trx,
es extremadamente versátil porque numerosas proteínas contienen dicho módulo
unido a otros dominios. Un panorama más detallado del conocimiento en este
campo se encuentra en varias revisiones recientes (8,9).
Por otra parte, algunas enzimas presentes en organismos multicelulares
catalizan la inserción de puentes disulfuro en las proteínas utilizando O2 y
liberando H2O2 [Prt-(SH)2 + O2 → Prt-(S)2 + H2O2] (10). Es destacable que
esta reacción particular no es totalmente independiente de las PDOR, por
cuanto el sitio catalítico de esta la familia de enzimas (i.e.
Quiescin-sulfhidril oxidasa) está constituido por un dominio Trx y un
dominio FAD.
Glutationilación de proteínas: Es evidente que el par redox GSSG/GSH tiene
un rol importante en los procesos de señalización mediante la modificación
de las cisteínas via el intercambio tiol-disulfuro. Utilizando este
mecanismo, una gran cantidad de proteínas tienen la capacidad de unir
reversiblemente el glutation al tiol de un residuo cisteína mediante la
formación de un heterodisulfuro [Prot-(S)2 + G-SH → HS-Prot-S−S-G] (11,12).
Esta modificación post-traduccional, denominada S-glutationilación, causa un
cambio funcional específico en la diana que es importante para la modulación
de los intermediarios implicados en los procesos de señalización.
Alternativamente, la S-glutationilación de las proteínas puede servir para
prevenir la oxidación de (i) las proteínas a S-oxiácidos (ver abajo), y/o (ii)
el GSH a GSSG.
Hay un amplio consenso en que las ROS y la regulación redox cumplen papeles
cruciales en procesos inflamatorios y en la respuesta inmune. En este
contexto, la glutationilación de las proteínas ha sido asociada a la
respuesta inflamatoria en enfermedades vinculadas con la inflamación crónica
como infecciones virales, diabetes, arterioesclerosis y cáncer. Los
mecanismos implicados no han sido meticulosamente caracterizados pero la
glutationilación afecta la actividad de numerosas proteínas que funcionan
como factores de transcripción, moléculas de adhesión, enzimas y citoquinas
(12). El mecanismo catalítico implicado en la formación de HS-Prot-S−S-G en
las
células es aún desconocido mientras que la escisión de este heterodisulfuro
es catalizada eficientemente por una PDOR particular que ha sido
caracterizada en la mayoría de los procariotes y eucariotes, i.e. la
glutarredoxina (13).
Formación de oxiácidos en los residuos cisteína de las proteínas
Los estudios recientes muestran claramente que la formación y ruptura de la
unión disulfuro no basta para explicar los múltiples efectos de las
proteínas dotadas de azufres reactivos. El átomo de azufre tiene una
configuración electrónica (3s23p4) que le permite adoptar estados de
oxidación entre +6 y -2. En las proteínas, los tioles de las cisteínas
constituyen el estado más reducido (estado de oxidación: – 2) pero la
capacidad del azufre para asumir múltiples estados de oxidación y formar
uniones con el oxígeno, el nitrógeno, los halógenos y el selenio origina un
rango de modificaciones post-traduccionales que están estrechamente
vinculadas a la señalización redox. El presente análisis está restringido a
los oxiácidos porque los nuevos experimentos muestran que los grupos
sulfénico [R-SOH], sulfínico [R-SO2H] y sulfónico [R-SO3H] juegan un rol
significativo en la remoción del H2O2 y en la señalización redox (Figura 5).
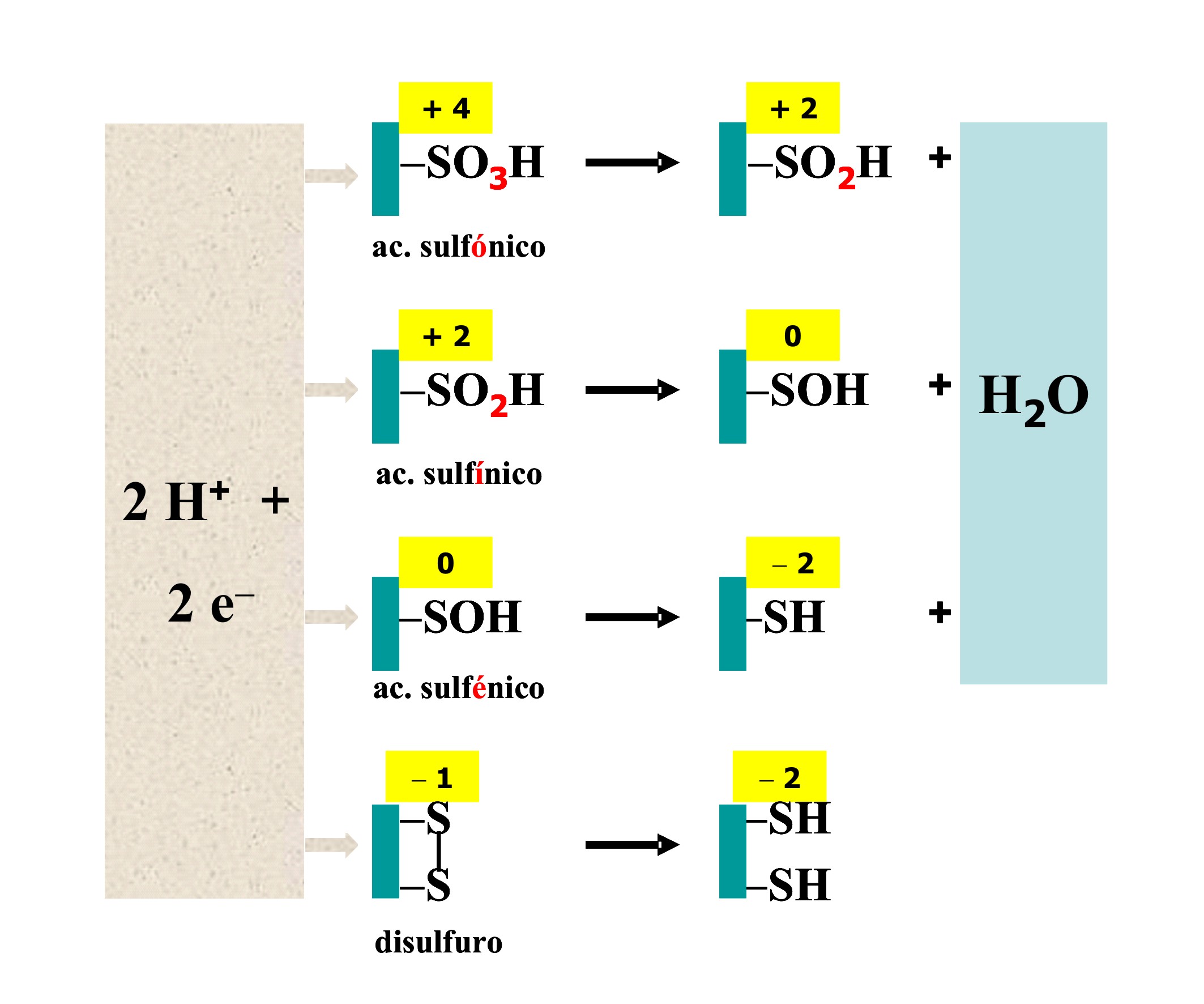
Figura 5. Reducción bielectrónica del átomo de azufre de las cisteínas.
Estas especies sobreoxidadas, cuyas cargas negativas imponen
nuevos requerimientos estéricos, pueden ser dañinas cuando afectan las
proteínas al azar. Sin embargo, la formación de oxiácidos puede favorecer el
plegamiento (estructura terciaria, la oligomerización (estructura
cuaternaria) y los procesos redox (función) cuando son manejadas
adecuadamente por procesos enzimáticos que implican cisteínas específicas.
La modificación de los grupos tiol en las proteínas con oxidantes tales como
H2O2 o peroxinitrito (ONO-O−) produce ácido sulfénico, el más simple
oxiácido de azufre orgánico. El microambiente que circunda las cisteínas
controla la reactividad de los tioles y la estabilidad de las modificaciones
determinando la dirección de las reacciones posteriores. El ácido sulfénico
puede condensar con otro tiol proteico o con GSH formando un puente
disulfuro cuya escisión retrotrae la proteína a su estado totalmente
reducido. Por ejemplo, el factor de transcripción sensor de estrés oxidativo
OxyR de E.coli y otras bacterias contiene una cisteína altamente reactiva
que forma no sólo un disulfuro intramolecular sino también ácido sulfénico
en respuesta al estrés oxidativo (14). Este sensor procariótico modula la
homeostasis redox activando la expresión de dos enzimas que remueven el
H2O2; i.e. la catalasa y la alquilhidroperóxido reductasa (AhpCF)(15). Sin
embargo, a menudo el R-SOH es vulnerable a sobreoxidaciones que conducen a
las formas más estables R-SO2H y R-SO3H. Esta capacidad dual del –Cys-SOH
para proceder en direcciones opuestas sugiere que la formación reversible
constituye un mecanismo adecuado por el cual los oxidantes celulares como
H2O2 pueden modular la catálisis enzimática, la homeostasis redox y los
eventos de señalización. Consistente con este concepto, las evidencias
acumuladas implican al ácido sulfénico no sólo en una amplia variedad de
procesos celulares sino también en numerosas patologías (16).
Una combinación de estudios estructurales y enzimáticos con la protein-tirosina
fosfatasa revelaron que el grupo sulfénico también puede reaccionar con el
nitrógeno de la unión peptídica generando la sulfenil-amide (sulfenamide)
(Figura 6) (17,18).
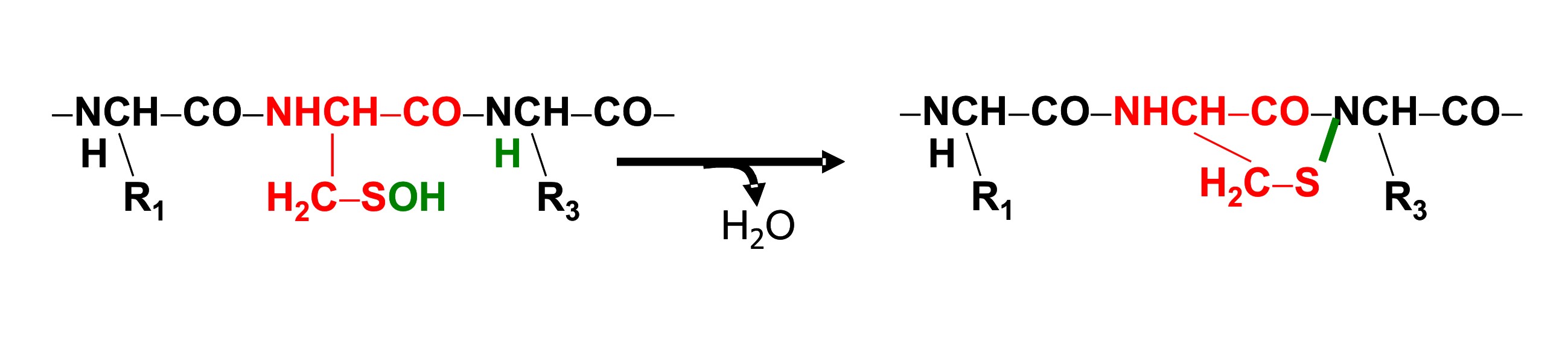
Figura 6. Formación del grupo sulfenamida intramolecular en
la protein-tirosina fosfatasa.
La presencia de esta estructura cíclica en la cadena
polipeptídica imparte un profundo cambio en el sitio activo de la protein-tirosina
fosfatasa regulando en consecuencia la interacción con otros dominios
proteicos (19,20). La recuperación de la protein-tirosina
NHCH−CO−H2C−SNHCH−CO−H2C−SNHCH−S
fosfatasa reducida por la acción del GSH sugiere que la formación de la
sulfenamida constituye un proceso acoplado a la señalización redox. [Nota:
la transformación de sulfénico a sulfenamida no implica un cambio en el
estado de oxidación del átomo de azufre]. Como la glutationilación, esta
modificación proteica inusual (i) contribuye a la regulación de la actividad
enzimática y (ii) protege las cisteínas previniendo la formación de ácido
sulfínico y sulfónico. Congruente con la ubicuidad de este mecanismo, la
proteína OhrR de Bacillus subtilis, un regulador transcripcional sensible a
peróxidos, contiene un grupo sulfenamida estable en solución y,
posiblemente, in vivo (14). En conjunto, estos nuevos estudios revelan que
el grupo sulfénico es una modificación post-traduccional con capacidades
regulatorias equivalentes a las del clásico intercambio tiol-disulfuro.
Regulación de la respuesta a ROS via las Enzimas Antioxidantes
La utilización de componentes citotóxicos como moléculas para la
señalización impone que la formación, la localización y la actividad las ROS
deben ser reguladas ajustadamente. Contrarrestando el desafío oxidativo, la
reacción espontánea con una batería de moléculas antioxidantes (e.g. GSH,
vitamina E, ascorbato, carotenoides, flavonoides) y un sistema enzimático
transforman las ROS en componentes biológicos menos agresivos. En este
aspecto, cuatro enzimas son muy eficientes en la transformación de las dos
ROS particulares, i.e. O2•−, H2O2. La superóxido dismutasa (2 O2•− + 2H+ →
H2O2 + O2) previene la interacción destructiva del anión superóxido con los
lípidos y las proteínas de membranas. Por otro lado, la transformación de
H2O2 implica la participación de tres familias de peroxidasas: catalasa,
glutation-peroxidasas y Prxs. La catalasa es una hemoproteína, generalmente
localizada en los peroxisomas, con actividad de H2O2-dismutasa (H2O2 → H2O +
½ O2) que eliminaría el H2O2 o prevendría su liberación hacia el exterior.
Alternativamente, las restantes peroxidasas catalizan la reducción del H2O2
utilizando el poder reductor de un dador de electrones (ROOH + 2 AH → ROH +
H2O + 2 A). En los mamíferos, es posible distinguir varias isoformas de las
glutation peroxidasas [H2O2 + 2 GSH → GSSG + 2 H2O] por la distribución
intracelular, pero una de ellas, generalmente citosólica y Se-dependiente,
reduce el H2O2 utilizando los electrones cedidos por el NADPH, via la
glutation reductasa [NADPH + H+ + GSSG → 2 GSH + NADP+]. En cambio, el H2O2
producido en las hojas de las plantas superiores es removido por el
sistema de la ascorbato peroxidasa [H2O2 + L-ascorbato → dehidroascorbato +
2 H2O] (21). Finalmente, las Prxs exhiben la capacidad peroxidasa para
transformar HO−OH, ONO−OH (peroxinitrito) y RO−OH (alquilhidroperóxidos).
Esta gran familia de proteínas ha recibido considerable atención en la
última década no sólo por controlar el tenor celular de H2O2 sino también
por participar activamente en el señalamiento redox (22).
Peroxirredoxinas
Las Prxs constituyen una gran familia de peroxidasas en la superfamilia de
proteínas que exhiben plegamiento de tipo Trx. La dificultad para definir el
rol que estas proteínas ubicuas juegan en la respuesta al estrés oxidativo
es la complejidad de isoformas. La gran diversidad en la estructura
primaria, las características de la estructura cuaternaria y el ciclo
catalítico para la reducción del hidroperóxido permitió agrupar esta familia
de proteínas en diversas subfamilias que se localizan en diferentes
compartimentos celulares (Tabla 2).
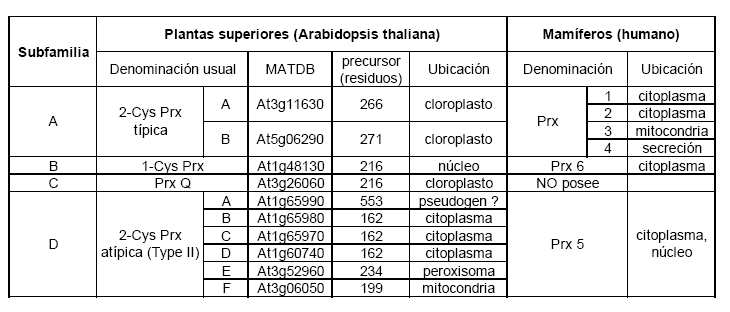
Tabla 2. Subfamilias de las Prxs en las plantas superiores y
en los mamíferos (23).
No todos los organismos vivos contienen todas las subfamilias
ni el mismo número de isoformas. Por ejemplo, Escherichia coli (una gamma
proteobacteria) contiene tres Prxs (24), Saccharomyces cerevisiae (levadura)
cinco (25), Homo sapiens (mamífero) seis (26,27) y Arabidopsis thaliana
(planta) diez (28).
Contrastando con los mayores sistemas de detoxificación de H2O2, que usan
grupos prostéticos unidos covalentemente al sitio activo, las Prxs emplean
el átomo de azufre de una cisteína conservada evolutivamente, denominada
cisteína peroxidática CysP, para la ruptura de la unión peroxilo –O–O– con
formación de un grupo sulfénico [–CysP-SH + H2O2 → –CysP-SOH + H2O]. La
presencia o ausencia de una segunda Cys, denominada cisteína resolutiva CysR
[–CysP-SOH + HS–CysR– → –CysP-S–S–CysR– + H2O], permite agrupar a las Prxs
en dos subfamilias: las 2-Cys Prx que conservan la Cys activa en la función
catalítica y las 1-Cys Prx que carecen de ella, requiriendo inevitablemente
un tiol externo para proseguir el ciclo catalítico. A su vez, la ubicación
del segundo tiol en la estructura primaria del polipéptido y la capacidad
para formar puentes disulfuro intercatenarios e intracatenarios permite
dividir a las 2-Cys Prx en “típicas” y “atípicas”, respectivamente (29,30).
2-Cys Prx típicas
La subfamilia de las 2-Cys Prx típicas muestra una importante conservación
de la estructura primaria en bacterias y eucariotas. Por ejemplo, la
estructura primaria de la 2-Cys Prx de los cloroplastos de colza es 52%
idéntica al ortólogo de mitocondrias de ratón. Dada la amplitud de funciones
relacionadas a esta subfamilia, en esta revisión enfatizaremos los aspectos
bioquímicos relevantes resaltando (i) la utilización de los estados de
oxidación del azufre en las cisteínas para modificaciones post-traduccionales
y (ii) las interacciones no-covalentes que inician respuestas apropiadas al
estrés oxidativo.
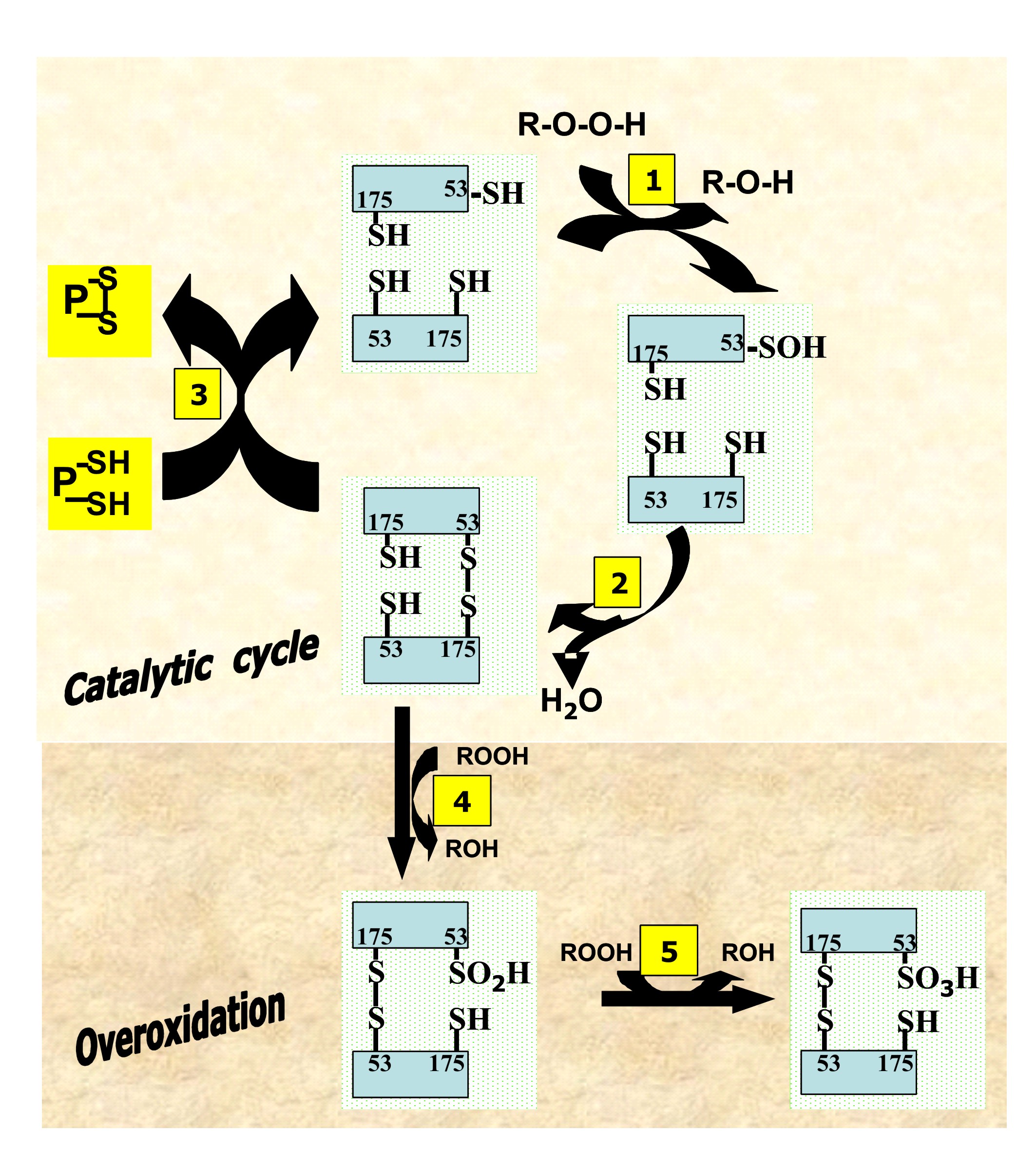
Figura 7. 2-Cys Prx. Rol de las cisteínas conservadas
evolutivamente en el ciclo catalítico y en la sobreoxidación.
Actividad peroxidasa: La actividad peroxidasa en la familia de las Prx comienza en la CysP ubicada en la region N-terminal de la cadena polipeptídica. Numerosos aminoácidos conservados alrededor de la CysP disminuyen el pKa del grupo tiol [–CysP-SH → –CysP-S− + H+] y, en consecuencia, estabilizan el anión tiolato para el ataque nucleofílico sobre el oxígeno terminal de la unión peroxilo (RO-OH). Este proceso genera el ácido sulfénico (R-SOH) en la CysP con la simultánea reducción del peróxido (Figura 7, reacción 1). La comparación de numerosos sitios proclives a la formación de –Cys–SOH en las proteínas reveló recientemente como varios factores (residuos polares, uniones hidrógeno) afectan la reactividad de los residuos cisteína (31). Luego de la formación del sulfenato en el sitio activo de las 2-Cys Prx, la reducción de la cisteína a la forma tiol ocurre en dos etapas (32). Primero, el sulfenato reacciona con el tiol localizado en la CysR de la subunidad complementaria formando un puente disulfuro intercatenario y liberando simultáneamente agua (Figura 7, reacción 2). Segundo, un sistema reductor complementario cierra el ciclo catalítico mediante un intercambio tiol-disulfuro con el par de cisteínas específicas de las PDOR (Figura 7, reacción 3). Uno de los rasgos bioquímicos interesantes del ciclo peroxidático es que al menos tres transformaciones redox diferentes tienen lugar en el sitio activo. Esencialmente, los tioles localizados en (1) la CysP, (2) la CysR, y (3) el motivo -CXXC- de la PDOR son los reductores para la formación sucesiva de ácido sulfénico en la CysP, el disulfuro intercatenario y el tiol en la CysP, respectivamente. Además, la primera y la última etapa implican la transferencia de hidrógenos (2H+ + 2 e−) mientras que la segunda, una deshidratación, constituye la reducción y la oxidación de la CysP y la CysR, respectivamente. En consecuencia, el átomo de azufre de la CysP pasa por tres diferentes estados de oxidación durante el ciclo peroxidático: tiol (-2) → ácido sulfénico (0) → disulfuro (-1) → tiol (-2).
Oxidación de la CysP: la oxidación de la CysP en la 2-Cys
Prx con oxidantes tales como el H2O2 y el peroxinitrito genera ácido
sulfénico, el más simple oxiácido del azufre orgánico. Este grupo tiene una
corta existencia por cuanto su reactividad lo hace vulnerable a la reducción
con tioles (Figura 7, reacción 2 y 3) o a las sobreoxidaciones que conducen
a los ácidos sulfínico y sulfónico (Figure 7, reacción 4 y 5). La naturaleza
transitoria de –CysP-SOH es adecuada para que los oxidantes celulares puedan
modular directamente la catálisis enzimática, la homeostasis redox y los
eventos de señalización celular. Consistente con esta idea, numerosos
estudios han implicado a –CysP-SOH en un amplia variedad de procesos
celulares y en numerosas patologías (16).
Reducción del sulfenato: La reactividad nucleofílica y electrofílica del
átomo de azufre en los sulfenatos facilita su reacción con otro grupo
sulfenato o con un tiol formando tiosulfinatos [–S–S(O)–] o disulfuros,
respectivamente (33,34). La primera reacción no ha sido encontrada en los
sistemas biológicos pero la segunda ocurre frecuentemente con formación de
puentes disulfuro vinculando dominios situados en la misma o en diferentes
cadenas polipeptídicas (15).
En caso particular de las 2-Cys Prxs típicas, el ácido sulfénico reacciona
con la CysR de la otra subunidad liberando agua y simultáneamente formando
un disulfuro intermolecular. Congruente con este mecanismo, la mutante de la
2-Cys Prx desprovista de la CysR remueve el H2O2 en presencia de un tiol
exógeno (e.g. ditiotreitol) mientras que la contraparte carente de la CysP
es completamente inactiva (35,36). Es decir, el tiol exógeno mantiene la
actividad peroxidasa porque reemplaza la CysR en el mecanismo que conduce a
la formación de un disulfuro con -CysP-SOH.
Reducción del disulfuro intermolecular: Una amplia variedad de PDOR y las
reductasas asociadas participan en la reducción que restituye los grupos
tioles a la CysP y a la CysR. El par Trx/NADP-Trx reductasa fue identificado
inicialmente en levaduras y mamíferos como el sistema que transporta el
poder reductor del NADPH hacia el disulfuro intercatenario de la 2-Cys Prx
(Figura 8)
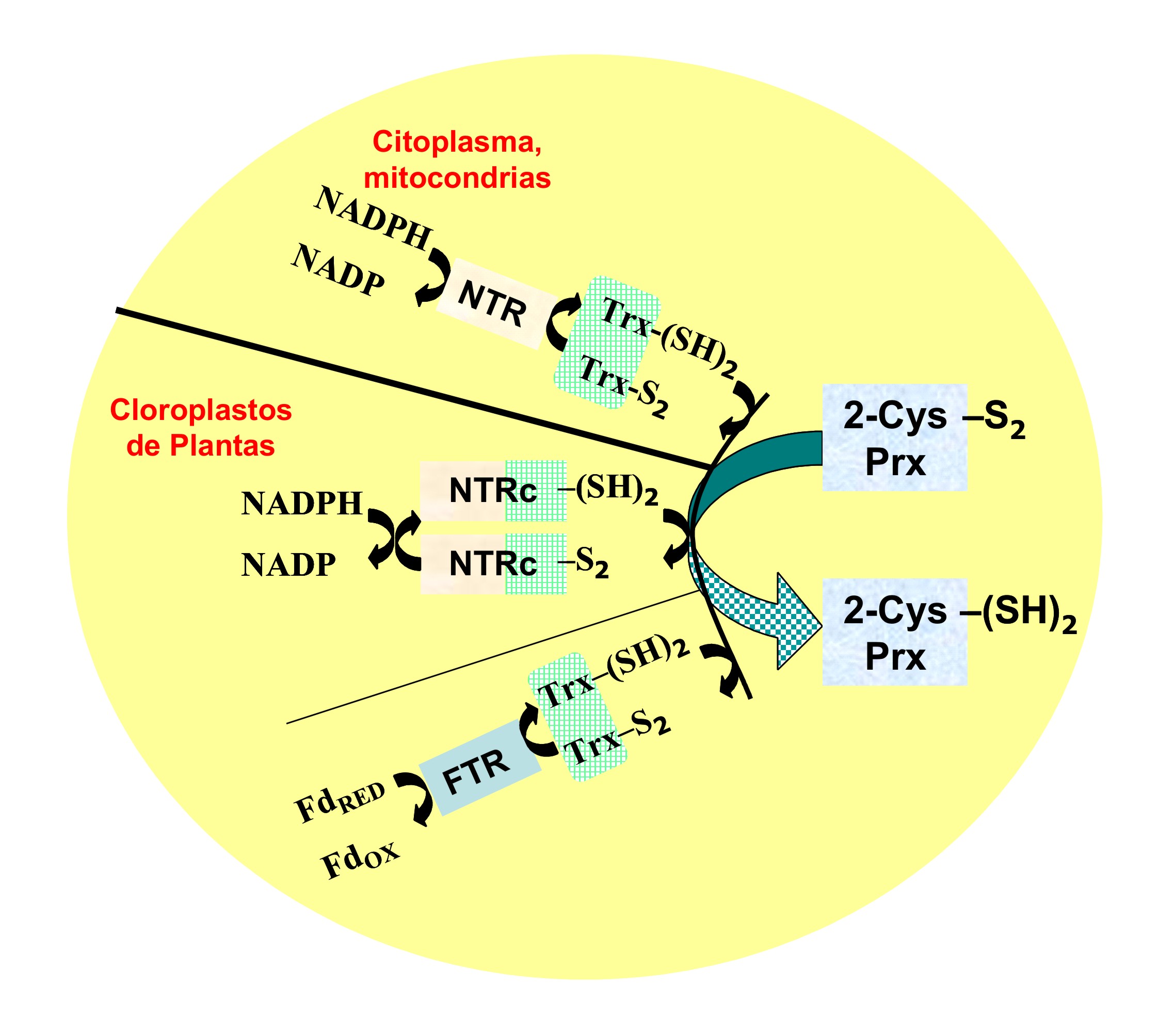
Figura 8. Reducción del puente disulfuro intercatenario de la
2-Cys Prx.
Sin embargo, los dadores fisiológicos de hidrógenos varían enormemente con el organismo, la ubicación intracelular, el estado de desarrollo y la respuesta a los estímulos ambientales. Por ello, no sorprende que los cloroplastos de las plantas superiores cuenten con dos sistemas para la reducción de la cistina intercatenaria: uno, que utiliza el NADPH via la NADP-Trx reductasa c (NTRc), y otro, que emplea la ferredoxina reducida generada por la luz en el sistema fotosintético via el par Trx/ferredoxina-Trx reductasa. Además, bacterias, tripanosomátidos y helmintos utilizan en esta función respectivamente a la alquilhidroperóxido reductasa (37), la triparredoxina (38), y el glutation (39). En este contexto, la capacidad de la Trx para reducir las 2-Cys Prx eucarióticas introduce un nivel mayor de complejidad por la multiplicidad de isoformas que estas PDOR exhiben en los diferentes seres vivos; e.g. tres isoformas en levaduras, dos en humanos y diecinueve en plantas (9).
Actividad Chaperona: El análisis bioquímico exhaustivo de
los caminos metabólicos ha revelado que numerosas proteínas cumplen más de
una función (40). El aspecto relevante de estas proteínas, denominadas “moonlighting
proteins”, es el uso de las modificaciones covalentes post-traduccionales y
las interacciones no-covalentes para alternar entre diferentes funciones y
responder en consecuencia a los cambios metabólicos. El estudio de las 2-Cys
Prxs de levaduras y humanas reveló que poseen una actividad chaperona que
está asociada a las transiciones en la estructura cuaternaria (41,42).
Estudios en procariotas y eucariotas mostraron que la 2-Cys Prx tiende a
formar ensamblados de alto peso molecular con pérdida de la actividad
peroxidasa y la simultánea aparición de la capacidad chaperona cuando las
células son expuestas a un estrés oxidativo o térmico (43,44). En línea con
cambios en las interacciones no-covalentes proteína-proteína, el estrés
oxidativo libera una 2-Cys Prx asociada a los ribosomas de levaduras
promoviendo su agregación (45). Dado que la 2-Cys Prx establece
interacciones no-covalentes con otras proteínas, sería esperable que algunas
funciones enzimáticas puedan ser ajustadas mediante dicho mecanismo.
Apoyando esta suposición, una de las enzimas claves para la asimilación
fotosintética de CO2, la fructosa-1,6-bisfosfatasa de los cloroplastos, es
estimulada por la 2-Cys Prx en un proceso que no utiliza la capacidad redox
de esta última (46).
Modificaciones post-traduccionales de la 2-Cys Prx: La
identificación de funciones adicionales en las 2-Cys Prx cambió la visión
clásica de un catalizador en la reducción de los hidroperóxidos a modulador
clave en importantes procesos biológicos. Dos mecanismos surgen para adecuar
la estructura de las 2-Cys Prx a las diferentes actividades: las
modificaciones post-traduccionales y las interacciones no-covalentes con
ligandos de bajo peso molecular. Fosforilación: La fosforilación de la 2-Cys
Prx humana en la treonina-90 provoca la formación de especies de alto peso
molecular que disminuyen marcadamente la actividad peroxidasa pero
incrementan la actividad chaperona (47). En línea con estos estudios, la
2-Cys Prx es fosforilada en una posición idéntica con reducción de la
actividad peroxidasa cuando los ratones son tratados con drogas que inducen
la enfermedad de Parkinson (48). Mientras que los residuos serina, treonina
y tirosina son fosforilados en la mayoría de las proteínas implicadas en la
transducción de señales, los grupos fosforilo unidos a residuos histidina,
aspártico y cisteína son menos frecuentes. En el último lustro, dos líneas
de investigación plantearon la fosforilación de la CysP y la CysR. Por un
lado, en S. cerevisiae se postula que la conversión de ácido sulfínico a
tiol implica la fosforilación de la CysP como un paso esencial (49). Por
otro lado, estudios recientes revelaron la incorporación de un grupo
fosforilo en las especies sobreoxidadas de la CysR, implicando a los
anhídridos mixtos fosforil-sulfínico [Prx–CysR–S(O)OPO32−] y
fosforil-sulfónico [Prx–CysR–S(O2)OPO32−] como una nueva alternativa para la
modificación post-traduccional de las proteínas (50). Actualmente no son
conocidos los roles de la supuesta fosforilación de la CysP y la
autofosforilación de la CysR pero evidentemente sugieren la factibilidad de
su participación en la transducción de señales. Es notable que la
incorporación covalente del grupo fosforilo en los oxiácidos de la 2-Cys Prx
integra en un único residuo la química no-redox del ATP con los múltiples
estados de oxidación del átomo de azufre (Figura 9) constituyendo un
mecanismo versátil para percibir cambios en los estados energéticos y redox
de la célula(23).
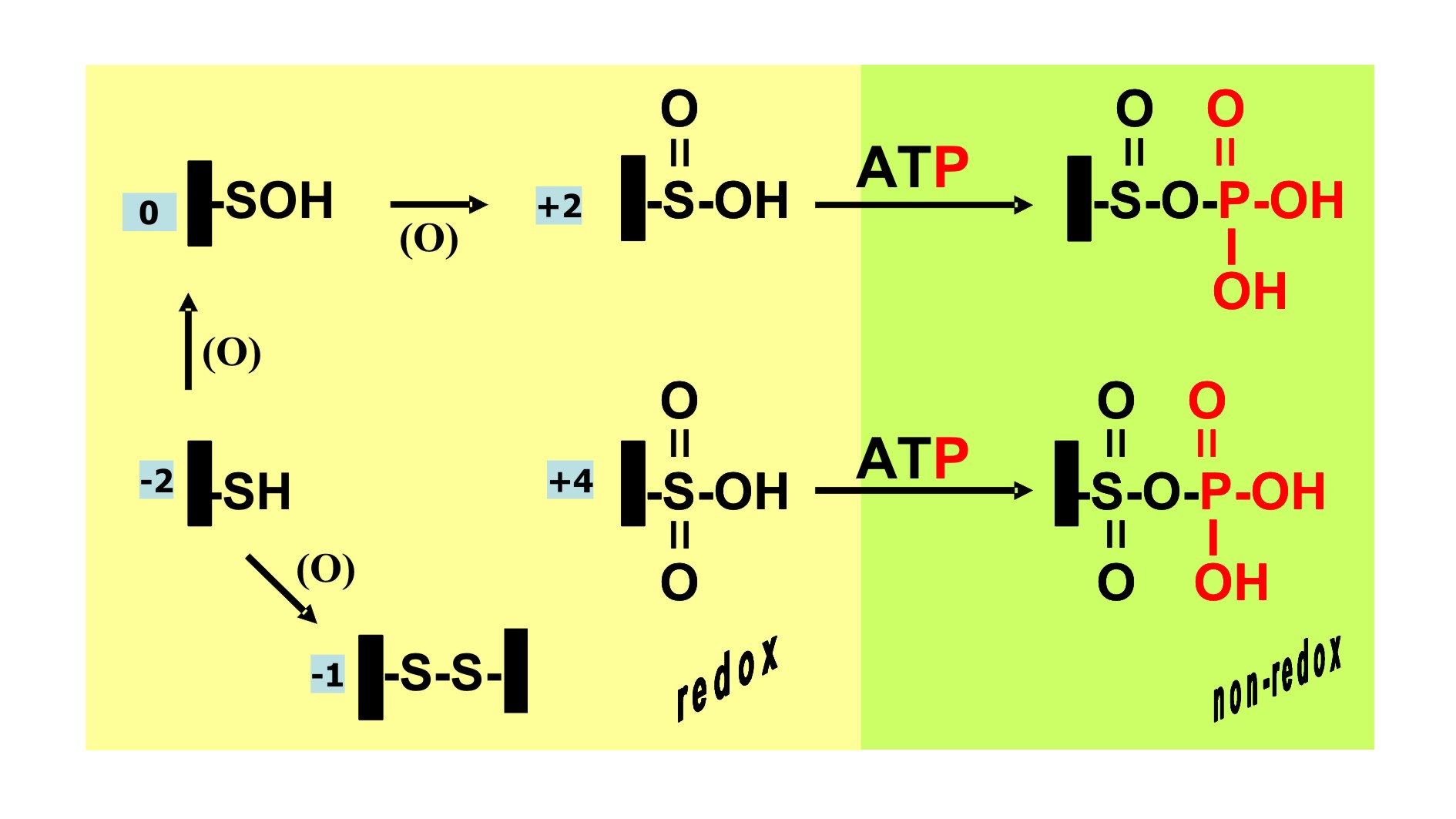
Figura 9. Química dual del átomo de
azufre en la CysR
Acetilación: Estudios recientes han mostrado que las dos
isoformas de la 2-Cys Prx humana están acetiladas en un residuo lisina
específico cuando líneas celulares humanas son (i) tratadas con el inhibidor
o (ii) desprovistas de una histona desacetilasa particular (51). Estos
resultados in vivo sugieren que la ausencia de una actividad desacetilasa
mantiene el grupo acetilo en la 2-Cys Prx. En línea con estos estudios, la
2-Cys Prx incorpora el grupo acetilo cuando es incubada in vitro con
acetil-CoA y la histona acetiltransferasa. Sorprendentemente, esta
acetilación específica incrementa no sólo la actividad peroxidasa sino
también la resistencia a la sobreoxidación.
Interacciones no-covalentes: Simultáneamente con el
hallazgo de la autofosforilación en la 2-Cys Prx fue observado que la acción
concertada del ATP y el Mg2+ inhibe la actividad peroxidasa pero sólo el
catión bivalente reduce la actividad chaperona (50). Estos estudios
revelaron (i) la regulación cinética de la actividad peroxidasa diferente
del control termodinámico dado por la disponibilidad del sustrato H2O2 y (ii)
el control diferencial de las actividades peroxidasa y chaperona por
reguladores desprovistos de capacidad redox. Experimentos complementarios
mostraron que la presencia de ATP y Mg2+ conduce la estructura cuaternaria
de la 2-Cys Prx a ensamblados de gran tamaño, los cuales retornan a la forma
inicial cuando son removidos los moduladores (M. Aran, resultados no
publicados). Estos estudios sugieren que la acción concertada de ATP y Mg2+
induce una variedad de especies de alto peso molecular, las cuales a su vez
generan nuevas funciones. Las proteínas dotadas de una alternancia en la
estructura cuaternaria que permite respuesta funcionales a la concentración
de proteínas y a los reguladores alostéricos han sido denominadas morfeínas
(52).
Conclusiones
La fisiología normal de las células, el envejecimiento y el estrés sistémico
o local generan ROS (53). Aunque estas especies reactivas han sido
consideradas tóxicas para los seres vivos, los estudios recientes sugieren
que tienen una importancia central en los procesos de señalización celular.
Esta peculiaridad requiere que, para regular los niveles intracelulares de
ROS, los organismos dispongan un sistema complejo que atempere los efectos
deletéreos y permita una respuesta adecuada a los estímulos. Los
antioxidantes modulan química y enzimáticamente el balance complejo entre
las
velocidades de formación de las ROS (estrés oxidativo) y la habilidad
celular para remover estas especies reactivas (capacidad antioxidante). De
esta manera, los antioxidantes reducen los niveles de ROS y determinan la
acumulación de las formas oxidadas en las proteínas sensoras –e.g. OxyR,
OhrR, Hsp33.
El cambio conceptual en la última década ha sido que las funciones
bioquímicas de los grupos tiol en las proteínas no sólo participan en la
formación de los puentes disulfuro sino también utilizan otros estados de
oxidación del átomo de azufre. En particular, el derivado sulfénico juega un
rol muy importante en los sitios catalíticos de las enzimas (protein-tirosina
fosfatasa), regula la actividad de factores de transcripción específicos (OxyR,
OhrR) y percibe el estrés oxidativo. La naturaleza transitoria y la
presencia en múltiples procesos sugiere fuertemente que el grupo sulfénico
en las cisteínas constituye un mecanismo por el cual las ROS pueden
desencadenar respuestas celulares.En este contexto, el estado de oxidación
del átomo de azufre (0) puede aumentar por sobreoxidación a los ácidos
sulfínico (+2) y sulfónico (+4) o disminuir por formación de un puente
disulfuro (-1), modificaciones que también estarían implicadas en
señalamiento, envejecimiento o patologías (54). Esta peculiaridad admite la
posibilidad de reconocer el estímulo y transducir paulatinamente la
respuesta con un único residuo de aminoácido, una notable ventaja respecto a
mecanismos “on/off” que requieren complejos sistemas de proteínas y/o
residuos para la misma función.
En este contexto, la formación de –Cys-SOH en las Prxs, y en particular las
2-Cys Prx, juega un rol importante en la percepción y regulación de los
estímulos ambientales. Esta familia de proteínas no sólo exhibe una amplia
diversidad de sitios de fosforilación y acetilación sino también puede ser
modulada alostéricamente por intermediarios metabólicos. Además de utilizar
al ATP para modular la estructura cuaternaria via transformaciones
covalentes e interacciones no-covalentes, la 2-Cys Prx posee la capacidad de
recurrir a la química no-redox para manejar situaciones de estrés oxidativo.
Sin embargo, es necesario tener en cuenta que la respuesta al desafío
oxidativo depende no sólo de la dosis y la duración del estímulo sino
también de la localización subcelular de la diana a ser protegida. Por ello
no sorprende que, en las plantas con metabolismo C4, el contenido de la
2-Cys Prx sea mayor en las células del mesófilo que en las células de la
vaina (55). En el futuro, el análisis detallado de los organismos
transgénicos establecerá el rol que los sistemas antioxidantes juegan en la
expresión temporal y espacial de los genes que participan en la respuesta a
los estímulos ambientales.
Referencias
1.- Jacob, C. Giles, G.I., Giles, N.M. and Helmut Sies, H. (2003) Sulfur and
Selenium: The Role of Oxidation State in Protein Structure and Function.
Angew. Chem. Int. Ed. 42, 4742-4758.
2.- Lee, J.W. and Helmann, J.D. (2006) The PerR transcription factor senses
H2O2 by metal-catalysed histidine oxidation. Nature 440, 363-367.
3.- Zhang, X.H. and Weissbach, X. (2008) Origin and evolution of the
protein-repairing enzymes methionine sulphoxide reductases. Biol. Rev. 83,
249-257.
4.- Winterbourn, C.C. and Hampton, M.B. (2008) Thiol chemistry and
specificity in redox signaling. Free Rad. Biol. Med. 45, 549-561.
5.- Cabiscol, E., Piulats, E., Echave, P., Herrero, E. and Ros, J. (2000)
Oxidative stress promotes specific protein damage in Saccharomyces
cerevisiae. J. Biol. Chem. 275, 27393-27398.
6.- DeYulia, G.J., Cárcamo, J.M., Borquez-Ojeda O., Shelton, C.C. and Golde,
D.W. (2005) Hydrogen peroxide generated extracellularly by receptor–ligand
interaction facilitates cell signaling. Proc. Natl. Acad. USA 102,
5044-5049.
7.- Triantaphylides, C., Krischke, M., Hoeberichts, F.M., Ksas, B., Gresser,
G., Havaux, M., Van Breusegem, F. and Mueller, M.J. (2008) Singlet Oxygen Is
the Major Reactive Oxygen Species Involved in Photooxidative Damage to
Plants. Plant Physiol. 148, 960-968.
8.- Buchanan, B.B. and Balmer, Y. (2005) Redox regulation: a broadening
horizon. Annu. Rev. Plant Biol. 56, 187–220.
9.- Mora-Garcia, S., Stolowicz, F. and Wolosiuk, R.A. (2005) Redox signal
transduction in plant metabolism. Control of primary metabolism in plants
(Plaxton, W. and McManus, M. eds.). Annu. Plant Rev. Vol.22. p. 150-186.
Blackwell Publishing. Oxford, UK.
10.- Thorpe, C., Hoober, K.L., Raje, S., Glynn, N.M., Burnside, J., Turi,
G.K., and Coppock, D.L. (2002). Sulfhydryl oxidases: emerging catalysts of
protein disulfide bond formation in eukaryotes. Arch. Biochem. Biophys. 405,
1-12.
11.- Dalle-Donne, I., Rossi, R., Colombo, G., Giustarini, D. and Milzani, A.
(2009) Protein S-glutathionylation: a regulatory device from bacteria to
humans. T. Biochem. Sci. 34, 85-96.
12.- Shelton, M.D. and Mieyal, J.J. (2008) Regulation by Reversible
S-Glutathionylation: Molecular Targets Implicated in Inflammatory Diseases.
Mol. Cells 25, 332-346.
13.- Lemaire, S.D. (2004) The glutaredoxin family in oxygenic photosynthetic
organisms. Photosynth. Res. 79, 305-318.
14.- Lee, J.W., Soonsanga, S. and Helmann, J.D. (2007) A complex thiolate
switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc.
Natl. Acad. Sci. USA 104, 8743-8748.
15.- Kiley, P.J. and Storz, G. (2004) Exploiting thiol modifications. PLoS
Biol. 2, e400.
16.- Poole LB, Karplus PA & Claiborne A (2004) Protein sulfenic acids in
redox signaling. Annu. Rev. Pharmacol. Toxicol. 44, 325-347.
17.- Salmeen, A., Andersen, J.N., Myers, M.P., Meng, T.C., Hinks, J.A.,
Tonks, N.K. and Barford, D. (2003) Redox regulation of protein tyrosine
phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769-773.
18.- van Montfort R.L.M., Congreve M., Tisi D., Carr R. and Jhoti H. (2003)
Oxidation state of the active-site cysteine in protein tyrosine phosphatase
1B. Nature 423, 773-776.
19.- Yang J., Groen A., Lemeer S., Jans A., Slijper M., Roe S.M., den Hertog
J. and Barford D. (2007) Reversible oxidation of the membrane distal domain
of receptor PTPalpha is mediated by a cyclic sulfenamide. Biochemistry 46,
709-719.
20.- Tonks N.K. (2006) Protein tyrosine phosphatases: from genes, to
function, to disease. Nature Rev. Mol. Cell Biol. 7, 833-846.
21.- Asada, K. (1999) The water–water cycle in chloroplasts: scavenging of
active oxygens and dissipation of excess photons, Annu. Rev. Plant. Physiol.
Plant. Mol. Biol. 50, 601-639.
22.- Rhee, S. G. (2006) Cell signaling: H2O2, a necessary evil for cell
signaling. Science 312,1882-1883.
23.- Aran, M., Ferrero, D.S., Pagano, E. and Wolosiuk, R.A. (2009) Typical
2-Cys peroxiredoxins. Modulation by covalent transformations and noncovalent
interactions. FEBS J. 276, 2478-2493.
24.- Zhou, Y., Wan, X.Y., Wang, H.L., Yan, Y.D., Hou, Y.D. and Jin, D.Y.
(1997) Bacterial Scavengase p20 Is Structurally and Functionally Related to
Peroxiredoxins. Biochem. Biophys. Res. Commun. 233, 848-852.
25.- Park, S.G., Cha, M.K., Jeong, W. and Kim, I.H. (2000) Distinct
Physiological Functions of Thiol Peroxidase Isoenzymes in Saccharomyces
cerevisiae. J. Biol. Chem. 275, 5723-5732.
26.- Zhou, Y., Kok, K.H., Chun, A.C., Wong, C.M., Wu, H.W., Lin, M.C.M.,
Fung, P.C.W., Kung, H.F. and Jin, D.Y. (2000) Mouse peroxiredoxin V is a
thioredoxin peroxidase that inhibits p53-induced apoptosis. Biochem. Biophys.
Res. Commun. 268, 921-927.
27.- Seo, M.S., Kang, S.W., Kim, K., Baines, I.C., Lee, T.H. and Rhee, S.G.
(2000) Identification of a new type of mammalian peroxiredoxin that forms an
intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 275,
20346-30354.
28.- Dietz, K.J., Horling, F., Konig, J. and Baier, M. (2002) The function
of the chloroplast 2-cysteine peroxiredoxin in peroxide detoxification and
its regulation. J. Exp. Bot. 53, 1321-1329.
29.- Knoops, B., Loumaye, E. and van der Eecken, V. (2007) Evolution of the
peroxiredoxins. In: Peroxiredoxin Systems. Structures and Functions,
Subcellular Biochemistry 44, edited by Flohé, L. and Harris, J.R. New York:
Springer, pp 27-40.
30.- Copley, S.D., Novak, W.R.P. and Babbitt, P.C. (2004) Divergence of
function in the thioredoxin fold suprafamily: Evidence for evolution of
peroxiredoxins from a thioredoxin-like ancestor. Biochemistry 43,
13981-13995.
31.- Salsbury Jr, F.R., Knutson, S.T., Poole, L.B. and Fetrow, J.S. (2008)
Functional site profiling and electrostatic analysis of cysteines modifiable
to cysteine sulfenic acid. Prot. Sci. 17, 299-312.
32.- Wood, Z.A., Schroder, E., Harris, J.R. and Poole, L.B. (2003) Structure,
mechanism and regulation of peroxiredoxins. T. Biochem. Sci. 28, 32-40.
33.- Allison, W. (1976) Formation and reactions of sulfenic acids in
proteins. Acc. Chem. Res. 9, 293-299.
34.- Claiborne, A., Mallett, T.C., Yeh, J.I., Luba, J. and Parsonage, D.
(2001) Structural, redox, and mechanistic parameters for cysteine-sulfenic
acid function in catalysis and regulation. Adv. Protein Chem. 58, 215-276.
35.- Chae, H.Z., Chung, S.J. and Rhee, S.G. (1994) Thioredoxin-dependent
peroxide reductase from yeast. J. Biol. Chem. 269, 27670-27678.
36.- Jara, M., Vivancos, A.P., Calvo, I.A., Moldon, A., Sanso, M. and
Hidalgo, E. (2007) The Peroxiredoxin Tpx1 is essential as a H2O2 scavenger
during aerobic growth in fission yeast. Mol. Biol. Cell 18, 2288-2295.
37.- Poole, L.B., Reynolds, C.M., Wood, Z.A., Karplus, P.A., Ellis, H.R. and
Calzi, M.L. (2000) AhpF and other NADH: peroxiredoxin oxidoreductases,
homologues of low Mr thioredoxin reductase. Eur. J. Biochem. 267, 6126-6133.
38.- Nogoceke, E., Gommel, D.U., Kiess, M., Kalisz, H.M. and Flohe, L.
(1997) A unique cascade of oxidoreductases catalizes trypanothione-mediated
peroxide metabolism in Crithidia fasciculata. Biol. Chem. 378, 827-836.
39.- Sayed, A.A. and Williams, D.L. (2004) Biochemical characterization of
2-Cys peroxiredoxins from Schistosoma mansoni. J. Biol. Chem. 279,
26159-26166.
40.- Jeffery, C.J. (2004) Molecular mechanisms for multitasking: recent
crystal structures of moonlighting proteins. Curr. Opin. Struct. Biol. 14,
663–668.
41.- Jang, H.H., Lee, K.O., Chi, Y.H., Jung, B.G., Park, S.K., Park, J.H.,
Lee, J.R., Lee, S.S., Moon, J.C., Yun, J.W. et al. (2004) Two enzymes in one;
two yeast peroxiredoxins display oxidative stress-dependent switching from a
peroxidase to a molecular chaperone function. Cell 117, 625–635.
42.- Moon, J.C., Hah, Y.S., Kim, W.Y., Jung, B.G., Jang, H.H., Lee, J.R.,
Kim, S.Y., Lee, Y.M., Jeon, M.G., Kim, C.W. et al. (2005) Oxidative stress-dependent
structural and functional switching of a human 2-Cys peroxiredoxin isotype
II that enhances HeLa cell resistance to H2O2-induced cell death. J. Biol.
Chem. 280, 28775–28784.
43.- Matsumura, T., Okamoto, K., Iwahara, S., Hori, H., Takahashi, Y.,
Nishino, T. and Abe, Y. (2008) Dimer–oligomer interconversion of wild-type
and mutant rat 2-Cys peroxiredoxin: disulfide formation at dimer–dimer
interfaces is not essential for decamerization. J. Biol. Chem. 283, 284–293.
44.- Chuang, M.H., Wu, M.S., Lo, W.L., Lin, J.T., Wong, C.H. and Chiou, S.H.
(2006) The antioxidant protein alkylhydroperoxide reductase of Helicobacter
pylori switches from a peroxide reductase to a molecular chaperone function.
Proc. Natl. Acad. Sci. USA 103, 2552-2557.
45.- Trotter, E.W., Rand, J.D., Vickerstaff, J. and Grant, C.M. (2008) The
yeast Tsa1 peroxiredoxin is a ribosomeassociated antioxidant. Biochem. J.
412, 73–80.
46.- Caporaletti, D., D’Alessio, A.C., Rodriguez-Suarez, R.J., Senn, A.M.,
Duek, P.D. and Wolosiuk, R.A. (2007) Nonreductive modulation of chloroplast
fructose-1,6-bisphosphatase by 2-Cys peroxiredoxin. Biochem. Biophys. Res.
Commun. 355, 722-727.
47.- Jang, H.H., Kim, S.Y., Park, S.K., Jeon, H.S., Lee, Y.M., Jung, J.H.,
Lee, S.Y., Chae, H.B., Jung, Y.J., Lee, K.O. et al. (2006) Phosphorylation
and concomitant
structural changes in human 2-Cys peroxiredoxin isotype I differentially
regulate its peroxidase and molecular chaperone functions. FEBS Lett. 580,
351–355.
48.- Qu, D., Rashidian, J., Mount, M.P., Aleyasin, H., Parsanejad, M., Lira,
A., Haque, E., Zhang, Y., Callaghan, S., Daigle, M. et al. (2007) Role of
Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s
disease. Neuron 55, 37–52.].
49.- Rhee S.G., Jeong W., Chang T.S. and Woo H.A. (2007) Sulfiredoxin, the
cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its
discovery, mechanism of action, and biological significance. Kidney Int.
Suppl. 106, S3-8.
50.- Aran, M., Caporaletti, D., Senn, A.M., Tellez de Iñon, M.T., Girotti,
M.R., Llera, A.S. and Wolosiuk, R.A. (2008) ATP-dependent modulation and
autophosphorylation of rapeseed 2-Cys peroxiredoxin. FEBS J. 275, 1450-1463.
51.- Parmigiani, R.B., Xu, W.S., Venta-Perez, G., Erdjument-Bromage, H.,
Yaneva, M., Tempst, P. and Marks, P.A. (2008) HDAC6 is a specific
deacetylase of peroxiredoxins and is involved in redox regulation. Proc.
Natl. Acad. Sci. USA 105, 9633–9638.
52.- Jaffe, E.K. (2005) Morpheeins. A new structural paradigm for allosteric
regulation. T. Biochem. Sci. 30, 490–497.
53.- Finkel, T. and Holbrook, N.J. (2000) Oxidants, oxidative stress and the
biology of ageing. Nature 408, 239-247.
54.- Cross, J.V. and Templeton, D.J. (2006) Regulation of signal
transduction through protein cysteine oxidation. Antioxid. Redox Signal. 8,
1819-1827.
55.- Majeran, W., Cai, Y., Sun, Q. and van Wijk, K.J. (2005) Functional
differentiation of bundle sheath and mesophyll maize chloroplasts determined
by comparative proteomics. Plant Cell 17, 3111-3140.
|
|
Revista QuímicaViva Número 3, año 8, Diciembre 2009 quimicaviva@qb.fcen.uba.ar |