La dolce vita: el papel de los azúcares en la biosíntesis de glicoproteínas
Dr. Julio J. Caramelo
Departamento de Química Biológica - FCEN -UBA
Instituto de Investigaciones Bioquímicas de Buenos Aires (CONICET)
Fundación Instituto Leloir
Av. Patricias Argentinas 435, Buenos Aires, C1405BWE, Argentina
e-mail: jcaramelo@leloir.org.ar
Resumen
El retículo endoplásmico es el lugar en donde se sintetizan las proteínas que ingresan a la vía secretoria. Previo a su salida, las proteínas adquieren su estructura terciaria y, de ser necesario, se ensamblan en oligómeros funcionales. Para facilitar estos procesos existen una gran variedad de chaperonas y enzimas facilitadoras del plegamiento. Asimismo, el estado de plegamiento es monitoreado por un sistema de control de calidad, el cual retiene en el retículo endoplásmico a aquellas especies que no han adquirido su conformación nativa. Si una proteína es incapaz de plegarse correctamente es retenida en el retículo endoplásmico y eventualmente es retrotranslocada al citosol para ser degradada por el proteasoma. Concomitantemente con el plegamiento tienen lugar diversas modificaciones postraduccionales, siendo las más destacadas la N-glicosilación y la formación de puentes disulfuro. Cerca de un cuarto de las proteínas de una célula eucarionte son N-glicosiladas, siendo la modificación postraduccional más frecuente. En las proteínas que alcanzaron su destino final los N-glicanos cumplen papeles fundamentales en diversos procesos de reconocimiento celular. Sin embargo los N-glicanos son utilizados también durante la maduración de las glicoproteínas como un sistema que codifica información acerca de su estado conformacional, siendo un elemento clave en varias instancias decisivas a lo largo de la vía secretoria. En este trabajo se presentan las diversas etapas que atraviesa una proteína desde su ingreso al retículo endoplásmico hasta su llegada a su destino final, poniendo especial atención a las funciones tempranas de los N-glicanos.
Palabras clave: BiP, calnexina, calreticulina, chaperona, control de calidad de plegamiento, glucosiltransferasa, N-glicosilación, oligosacaril transferasa, proteína disulfuro isomerasa, retículo endoplásmico.
La dolce vita: the role of sugars on the biosynthesis of glycoproteins
Abstract
The proteins that enter the secretory pathway are synthesized in the endoplasmic reticulum. Previously to their exit from the endoplasmic reticulum, the proteins acquire their tertiary structure and, if necessary, they form oligomeric structures. These processes are monitored by a folding quality control system. Those species unable to fold properly are retained in the endoplasmic reticulum and, eventually, are retrotranslocated into the cytosol for their degradation by the proteasome. Along the folding process, several postranslational modifications take place, being the most important the addition of N-glycans and the formation of disulfide bridges. About one quarter of the eucariotic proteins are N-glycosylated: this is the most common postranslational modification. The N-glycans are fundamental in many molecular recognition events once a protein has reached it final destination. In addition, the N-glycans are also employed as a system that encodes information about the conformational status of a glycoprotein, playing a central role in many decisive points along the life of a glycoprotein. Here I present the journey of a protein form the endoplasmic reticulum to its final destination, focusing the attention on the early roles played by the N-glycans.
Keywords: BiP, calnexin, calreticulin, chaperone, endoplasmic reticulum, folding quality control, glucosyltransferase, N-glycosylation, oligosaccharyltransferase, protein disulfide isomerase, ,.
Abreviaturas: BiP, proteína que une la cadena pesada de las inmunoglobulinas (la HSP70 del RE), CRT, calreticulina; CNX, calnexina; GRP94, proteína regulada por glucosa de 94 kDa (la HSP90 del RE); GT, UDP-Glc:glicoproteína glucosiltransferasa; PDI, proteína disulfuro isomerasa; RE, retículo endoplásmico
Introducción
El retículo endoplásmico (RE) es un sistema intracelular de compartimientos que abarca cerca del 10 % del volumen de un célula eucarionte. Esta organela cumple al menos tres funciones:
a) Segundo sitio de síntesis de proteínas, luego del citosol.
b) Principal lugar de síntesis de lípidos.
c) Principal reservorio intracelular de calcio en células no contráctiles.
Las proteínas que ingresan al RE perteneces a lo que se denomina vía secretoria. Estas proteínas comprenden cerca de un tercio del genoma de una célula eucarionte y tienen como destino final ciertas organelas (RE, aparato de Golgi o lisosomas), la membrana plasmática o la secreción al medio extracelular. La gran diversidad de receptores, transportadores y canales presentes en la membrana plasmática dan cuenta en gran medida de su abundancia en el genoma. En algunos tipos celulares especializados en secreción (linfocitos B activados, células pancreáticas beta, etc.) el RE comprende la mayor parte del volumen celular, y la maquinaria de síntesis proteica está dedicada principalmente a sintetizar unas pocas proteínas. Cuando una proteína ingresa al RE, ya sea co- o postraduccionalmente, lo hace a través de un complejo denominado translocón, cuyo componente central es una proteína integral de membrana llamada SEC61 (1). Esta proteína forma un poro estrecho por el cual ingresan las proteínas en forma desplegada. Luego de ingresar al RE, las proteínas adquieren su estructura terciaria, y de ser necesario se ensamblan en complejos oligoméricos. Estos procesos son supervisados por diversos sistemas de control de calidad, los cuales evitan la salida de aquellas proteínas que no han adquirido su conformación nativa. Las proteínas incapaces de plegarse eventualmente son retrotranslocadas al citosol, en donde son degradadas por el proteasoma en un proceso denominado ERAD (Endoplasmic Reticulum Associated Degradation). Concomitantemente con el plegamiento y ensamblado tienen lugar una serie de modificaciones post- y cotraduccionales, siendo las más importantes la formación de puentes disulfuro y la N-glicosilación.
Plegamiento de proteínas en el RE
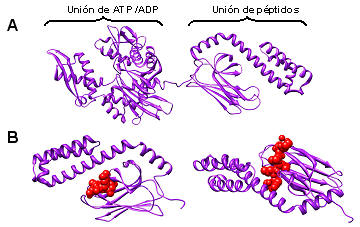
El RE dispone de una enorme variedad de chaperonas y enzimas que asisten a la maduración conformacional de las proteínas. Entre las chaperonas clásicas se destacan BiP y Grp94, las versiones del RE de HSP70 y HSP901, respectivamente (2-4). BiP es la proteína más abundante de esta organela, dando cuenta de aproximadamente el 5 % del total. Esta chaperona reconoce péptidos hidrofóbicos de unos 7 residuos de longitud, los cuales se unen en un proceso acoplado a la hidrólisis de ATP (5). Para cumplir con su función BiP cuenta con cochaperonas que la asisten en la unión de los sustratos (SEC63 y ERdj3, que son homólogas a HSP40) y en el intercambio de ADP por ATP (denominadas NEFs, por nucleotide exchange factor). Prácticamente todas las proteínas sintetizadas en el RE son asistidas en su plegamiento por BiP. Cuando las proteínas ingresan al RE usualmente son recibidas por BiP. Para ello son presentadas por SEC63, una homóloga de HSP40 anclada a la membrana del RE que se encuentra asociada al translocón. Se han resuelto varias estructuras del dominio de unión a péptidos y de unión a ATP, y recientemente apareció la primer estructura de la DnaK de E. coli completa (Fig. 1A). El sitio de unión a péptidos está formado por una hoja beta, el cual está cubierto por una hélice alfa que funciona como tapa. Esta disposición obliga que los péptidos adopten una conformación extendida para poder unirse a estas chaperonas (Fig. 1B). La estructura de BiP se predice que debería ser muy similar a la de DnaK.
1HSP70 y HSP90 significa "heat shock protein" de 70 y 90 kDa, respectivamente.
Figura 1. (A) Estructura de DnaK con sus dos dominios (PDB: 2KHO). (B) Dos vistas del dominio de unión de péptidos formando un complejo con el péptido NRLLLTG (PDB: 1DKZ).
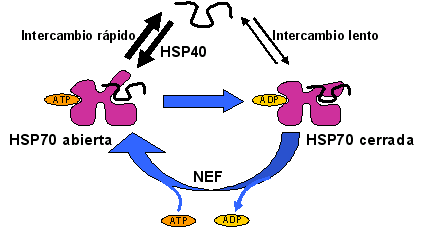
Las HSP70 pueden adoptar dos conformaciones. En el estado que une ATP el dominio de unión a péptidos presenta una conformación "abierta", en donde la tapa está distanciada de la hoja beta. En esta conformación la velocidad de entrada y salida del sustrato es rápida. La unión de los péptidos gatilla la hidrólisis del ATP y como consecuencia la chaperona sufre un cambio conformacional hacia una forma "cerrada" que atrapa al sustrato (es la mostrada en la Fig. 1B). El mecanismo por el cual la unión del sustrato induce la hidrólisis de ATP, es decir cómo se acoplan las actividades de los dominios, no es claro. A continuación se intercambia el ADP por ATP, la proteína vuelve a la conformación abierta y el péptido es liberado (Fig. 2).
Figura 2. Ciclo catalítico de la HSP70
Si bien las HSP70 son fundamentales para la maduración conformacional de muchas proteínas, su mecanismo de acción es llamativamente simple. En principio se limitan a unir y soltar segmentos hidrofóbicos en un proceso acoplado a la hidrólisis de ATP. Su efectividad para favorecer el plegamiento es indirecta, y es principalmente una consecuencia de prevenir la agregación de proteínas mal plegadas. Esto se debe a que mientras la adquisición de la estructura terciaria es un proceso unimolecular, en la formación de agregados intervienen varias proteínas y su velocidad se incrementa notablemente con la concentración. La unión de los intermediarios de plegamiento a la HSP70 disminuye su concentración como especies libres, inhibiendo entonces la agregación.
A diferencia de la HSP90 citosólica, que cumple funciones reguladoras, la principal función de Grp94 a asistir al plegamiento (6). El rango de sustratos de esta chaperona es más reducido que el de BiP, y en general actúa durante etapas avanzadas del plegamiento. No conocemos qué reconoce Grp94 en sus sustratos. Se especula que serían parches de aminoácidos hidrofóbicos expuestos en la superficie. Por otra parte, Grp94 une grandes cantidades de calcio con baja afinidad, siendo uno de los dos buffer de calcio más importantes del RE (el otro es la calreticulina). Llamativamente en el RE no se ha encontrado ninguna proteína de la familia de las chaperoninas (TRIC en el citosol de eucariontes, GroEL en bacterias). Esto puede deberse a que algunas de las funciones que normalmente llevan a cabo las chaperoninas en el citosol podrían ser suplidas en el RE por Grp94.
La principal función de BiP y Grp94 es evitar las interacciones inespecíficas entre los polipéptidos, de forma que previenen la formación de agregados. Asimismo, al ser proteínas residentes del RE, estas chaperonas evitan que las proteínas unidas a ellas prosigan su ruta por la vía secretoria. Es decir que una proteína con defectos estructurales será retenida en el RE. Si una proteína es incapaz de adquirir su conformación nativa, por ejemplo por tener una mutación que afecte su plegamiento, eventualmente se convierte en sustrato del ERAD. Si bien inicialmente se postuló que la degradación de proteínas de la vía secretoria ocurría en los lisosomas, hoy sabemos que este proceso se lleva a cabo principalmente por el proteasoma en el citosol. Para ello las proteínas deben ser retrotranslocadas, en un proceso que comprendemos muy parcialmente y en el que intervienen numerosos componentes (7). Durante la retrotranslocación las proteínas son desplegadas, y si bien originalmente se postuló que atravesaban la membrana a través del SEC61, hoy sabemos que no es así y han surgido otras proteínas como posibles canales (DERLIN 1, 2 y 3). Del lado citosólico la proteína es recibida por diversos componentes, entre los que se destacan una E3 ubiquitin ligasa asociada a la membrana del RE, una AAA+ ATPasa soluble del citosol (p97) que ayudaría a sacar el sustrato del RE, y una N-glicanasa que corta los N-glicanos de las glicoproteínas antes de su ingreso al proteasoma.
Respuesta a las proteínas mal plegadas (UPR)
Al acumularse proteínas mal plegadas se activan diversos mecanismos tendientes a restaurar la homeostasis celular, en un proceso denominado unfolded protein response (UPR). La UPR modula la capacidad biosintética de la célula de formas muy variadas. Tres proteínas de membrana residentes en el RE funcionan como sensores de estrés: PERK, ATF6 e IRE1 (8). Las levaduras sólo cuentan con IRE1, mientras que los metazoos disponen de los tres sensores. El mecanismo para detectar la acumulación de proteínas mal plegadas es similar en los tres casos (9). En condiciones de reposo estas proteínas se encuentran unidas a BiP, quien las mantiene inactivas. Cuando se acumulan proteínas mal plegadas BiP se une a ellas, desplazándose de su complejo con los sensores (Fig. 3). Este mecanismo de titulación ha sido demostrado de diversas maneras, sin estar del todo claro la concentración de proteínas desplegadas y el grado de perturbación estructural necesarios para iniciar la respuesta.
El primer sensor que se activa es PERK, quien frena la traducción general de proteínas. El sentido de esta respuesta inicial es darle tiempo a las chaperonas para que recuperen las proteínas mal plegadas que se acumularon. El apagado de la traducción sucede bastante rápido (aproximadamente 30 minutos) y usualmente se reinicia al cabo de una o dos horas. PERK es una proteína quinasa que al disociarse de BiP dimeriza, se autofosforila y luego fosforila al factor de elongación eIF2a. Esto inhibe el ensamblado del complejo de iniciación de la traducción. Sin embargo existen algunos mRNAs (ATF4, ATF3, etc) que al fosforilarse eIF2a aumentan su traducción, por presentar marcos de lectura abiertos cerca del extremo 5´ de su mensajero. En condiciones normales estos marcos de lectura son leídos eficientemente, de forma que el ribosoma saltea el marco de lectura que se encuentra río abajo. Al fosforilarse eIF2a disminuye la eficiencia en la iniciación de la traducción, y en estas condiciones se puede llegar a leer los marcos que se encuentran río abajo. Eventualmente eIF2a es defosforilado por uno de estos productos, la primera parte de la UPR se apaga y la célula se prepara para las etapas subsiguientes.
A continuación se activan mecanismos que llevan a: (i) la síntesis de más chaperonas, (ii) la activación de la máquina de degradación y, (iii) la síntesis de membrana del RE. Esta última respuesta se cree que incrementa el volumen del RE, de forma de "diluir" las proteínas mal plegadas y dar mas "espacio" para las chaperonas (esta interpretación puede ser demasiado simple). El sensor que se activa a continuación de PERK es ATF6. Esta proteína posee señales de localización en el Golgi que se encuentran ocultas por BiP, de forma que en condiciones de reposo el complejo se localiza en el RE. Al acumularse proteínas mal plegadas y despegarse de BiP, ATF6 migra al Golgi en donde es cortada por dos proteasas (S1P y S2P) que liberan el dominio citosólico de la misma. Este dominio es un factor de transcripción que activa la síntesis de chaperonas y de XBP1, producto necesario para la siguiente etapa de la UPR. Finalmente el último efector en activarse es IRE1. Esta proteína, de forma similar a PERK, al liberarse de BiP dimeriza, se autofosforila y se prende su actividad de endoribonucleasa. IRE1 cataliza la escisión de un intrón en un mRNA que codifica para el factor de transcripción XPB1. Este proceso de corte y empalme genera una proteína XBP1 con un extremo C-terminal alternativo, la cual presenta actividad como factor de transcripción de numerosos genes que codifican para chaperonas y componentes del ERAD.
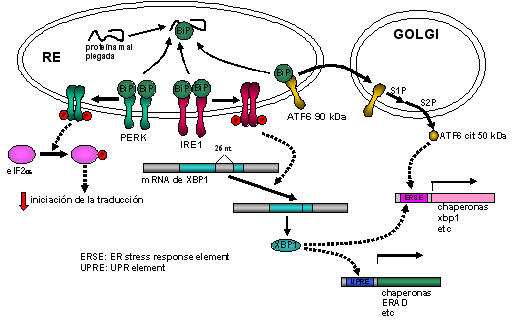
Figura 3. Las tres vías de activación de la UPR.
Estas cascadas de señalización están finamente reguladas, de forma que en determinadas situaciones algunas vías puede no prenderse. Es importante mencionar que la acumulación de proteínas mal plegadas no necesariamente implica un proceso patológico. Si bien en el laboratorio la UPR se induce mediante agentes farmacológicos (tapsigargina, tunicamicina, DTT, etc), la UPR también se activa durante procesos de desarrollo y diferenciación celular, los cuales demandan una mayor síntesis de proteínas. Un ejemplo clásico de aumento de demanda de la vía exocítica es la diferenciación de los linfocitos B en células plasmáticas, las cuales secretan miles de moléculas de anticuerpos por segundo. Durante este proceso se activan IRE1 y ATF6, y no se prende la vía mediada por PERK (10). Esto tendría lógica, dado que no tiene sentido frenar la traducción justamente cuando se necesita sintetizar proteínas.
Formación de puentes disulfuro
Topológicamente el lumen del RE es equivalente al medio extracelular. Una de las diferencias más notables que presenta respecto del citosol es un potencial redox más oxidante, de forma que la oxidación de cisteínas para formar puentes disulfuro es un proceso favorable. Cuando una proteína ingresa al RE presenta sus cisteínas en forma reducida. Aquellas proteínas con más de dos cisteínas tienen la posibilidad de formar más de un puente disulfuro. Por ejemplo, una proteína con 8 cisteínas que dan lugar a 4 puentes disulfuro dispone de 105 combinaciones posibles para formarlos. En este caso, si el proceso de oxidación fuera azaroso tendríamos que menos del 1 % de las proteínas presentarían la combinación adecuada de puentes disulfuro. De no mediar ningún mecanismo que corrija esta situación la mayor parte de estas moléculas serían inactivas, lo cual implicaría un derroche enorme de energía. En la evolución han surgido una familia de enzimas denominadas proteína disulfuro isomerasas (PDI) encargadas de resolver este problema (11).
Las PDIs poseen en su secuencia al menos un dominio homólogo a la tioredoxina, el cual presentan el motivo conservado C-X-Y-C (12). Una de estas cisteínas se encuentra oculta en el interior de la proteína y se la denomina "resolutiva", mientras que la otra está expuesta en la superficie. Estos residuos pueden formar un puente disulfuro entre sí, o alternativamente la cisteína expuesta puede formar un puente disulfuro mixto con otra proteína. Por ejemplo la PDI de Saccharomices cerivisiae tiene cuatro dominios homólogos a tioredoxina en el orden a-b-b´-a´ (Fig. 4). Los dominios a y a´ presentan el motivo CGHC y son activos, mientras que los dominio b y b´ no tienen este motivo.
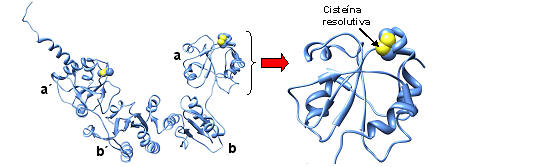
Figura 4. Estructura cristalográfica de la proteína disulfuro isomerasa de S. cerevisiae (PDB: 3BOA). Izquierda: detalle del dominio a en donde se muestran las cisteínas del sitio activo.
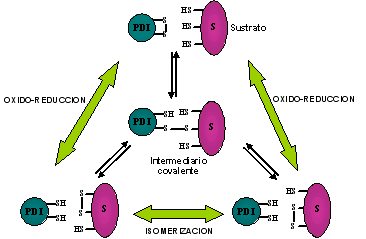
Las PDIs presentan actividad de oxidoreductasa y de disulfuro isomerasa (Fig. 5). Mediante la primera actividad generan los puentes disulfuro. En este proceso la PDI en forma oxidada se reduce al formar un puente disulfuro mixto con una cisteína del sustrato. Este puente se abre en un paso subsiguiente mediante la formación de un puente disulfuro interno del sustrato, quedando la PDI en forma reducida. El resultado de este proceso es la transferencia de un puente disulfuro desde la PDI al sustrato. La PDI reducida es vuelta a oxidarse mediante un sistema de reciclado en donde el último componente es una enzima denominada Ero1p, la cual transfiere los electrones al oxígeno. Alternativamente aquellos sustratos que ya formaron puentes disulfuro pueden ser reducidos por la PDI para generar un complejo covalente, el cual es resuelto por algún otro residuo de cisteína del sustrato que no formaba parte del puente original. El resultado de este proceso de reducción y posterior oxidación es la isomerización de los puentes disulfuro dentro del sustrato.
Figura 5. Actividades de las PDIs.
Desconocemos si las PDIs "saben" cuando el sustrato formó los puentes disulfuros correctos y por ende deben frenar su actividad. Aparentemente existe un juego entre estabilidad y cinética, en donde los puentes disulfuro "nativos" serían menos susceptibles a ser reducidos por las PDIs. En el retículo de una célula eucarionte hay al menos unas 18 PDIs diferentes. En algunos casos se ha podido identificar que ciertas PDI presentan especificidad por algunos sustratos, por ejemplo la ERp57 es una PDI dedicada a la isomerización de puentes disulfuro de glicoproteínas solubles (13). Sin embargo, todavía estamos lejos de comprender la gran complejidad de este sistema.
N-Glicosilación
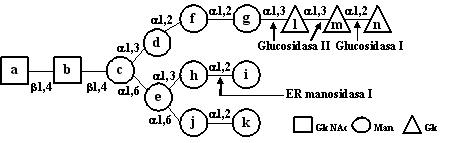
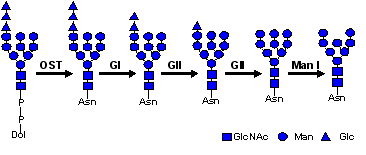
La N-glicosilación es una modificación en donde el oligosacárido GlcNAc2Man9Glc3 se une covalentemente a un residuo de asparagina mediante una unión N-glicosídica (Fig. 6). Casi todos los eucariontes transfieren este oligosacárido, con la excepción de algunos tripanosomástidos que transfieren GlcNAc2Man9. El residuo de asparagina modificado forma parte del secuón2 Asn-X-Ser/Thr, en donde X puede ser cualquier residuo excepto prolina. Asimismo existen casos en donde la secuencia Ans-X-Cys puede ser empleada.
Figura 6. Estructura del oligosacárido transferido en el RE y sitios de corte de las enzimas encargadas del procesamiento inicial
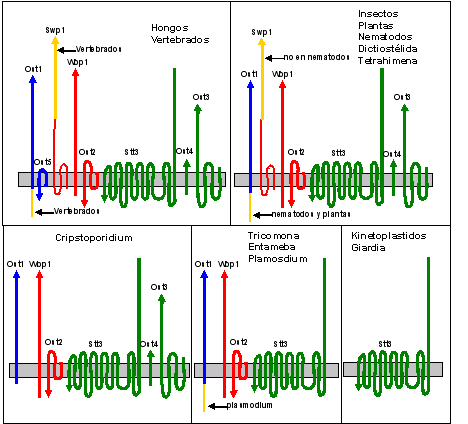
El oligosacárido es transferido en bloque desde dolicol-pirofosfato por un complejo llamado oligosacariltransferasa (OST), el cual en vertebrados está compuesto por ocho subunidades diferentes (OST1, OST2, OST3, OST4, OST5, STT3, Swp1 y Wbp1) (14). STT3 es la subunidad catalítica, y es la única subunidad presente en giardia y en los kinetoplástidos (Fig. 7). No es claro el rol de las demás subunidades, aunque se supone que regulan la especificidad de la transferencia (15). Este complejo se encuentra asociado al translocón, de forma que la glicosilación tiene lugar cotraduccionalmente. La presencia de un sitio de glicosilación no garantiza que el mismo vaya a ser modificado y cerca del 85 % de los sitios posibles son utilizados. En este sentido, se ha observado que la tendencia local a formar estructuras secundarias puede modular la eficiencia de la OST. Dado que aproximadamente un tercio de las proteínas de un eucarionte ingresan a la vía secretoria, tenemos que cerca de un cuarto de las proteínas totales son N-glicosiladas, siendo la modificación postraduccional más frecuente (16).
2Secuón: secuencia conservada de aminoácidos que es modificada químicamente.
Figura 7. Subunidades de la OST presentes en los diversos eucariontes
Si bien inicialmente se postuló que la N-glicosilación ocurre siempre cotraduccionalmente, evidencia reciente indica que algunas proteínas pueden ser glicosiladas luego de haber finalizado su síntesis. Llamativamente en vertebrados, plantas e insectos existen dos isoformas de la subunidad catalítica de la OST (STT3A y STT3B). Actualmente se postula que la STT3A estaría dedicada a la N-glicosilación cotraduccional, mientras que la STT3B sería responsable de la N-glicosilación postraduccional (17). Desconocemos la relevancia biológica de estas observaciones, sin embargo la existencia de dos isoformas de STT3 sería una forma de asegurarse una mayor ocupación de los secuones de glicosilación.
Funciones de los N-Glicanos
La N-glicosilación cumple diversos roles (18). Por un lado, los glicanos son componentes esenciales en procesos de reconocimiento célula-célula y célula-matriz, modulando fenómenos de adhesión, secreción y diferenciación. La gran variedad estructural que pueden presentar los azúcares y la posibilidad modificar el perfil químico de un glicano a los largo de la vida celular las convierten en moléculas ideales para modular estos procesos. Sin embargo, estas funciones, que podríamos llamar "fisiológicas", están superpuestas con efectos directos que ejercen sobre la estructura de las glicoproteínas. Los glicanos son moléculas hidrofílicas de gran tamaño que pueden modular el plegamiento y la estabilidad de las proteínas que modifican. Un glicano de alta manosa pesa unos 2000 Da, mientras que uno tetrantenario polisialilado puede pesar unos 5000 Da. Claramente es una modificación química bastante más notoria que muchas de las modificaciones postraduccionales más comunes (fosforilación, acetilación, metilación, etc.). En cuanto a la modulación de la estabilidad proteica los efectos son variados (19). En muchos casos la presencia de un N-glicano aumenta la estabilidad térmica de una proteína y disminuye la susceptibilidad a proteasas. Sin embargo, en otros tantos casos la estabilidad de una glicoproteínas no se ve afectada luego de desglicosilarla. Por otra parte, en general los glicanos disminuyen la tendencia a formar agregados proteicos, aumentado marcadamente la reversibilidad de los procesos de desnaturalización. Este puede adscribirse a un efecto de "protección hidrofílica", en donde el glicano cubre la superficie proteica previniendo las interacciones inespecíficas, comportándose como si fueran chaperonas químicas. A nivel de la estructura local, la presencia de un glicano favorece la formación de giros en la cadena polipeptídica (20), y muy comúnmente se los encuentra en este tipo de estructuras. Finalmente, algunos glicanos pueden no tener efectos una vez que la proteína adquirió su estructura nativa pero son necesarios para asistir en su maduración conformacional en el RE. Esta gran variedad de funciones dificulta la asignación de un rol específico a un dado glicano. La ausencia de actividad biológica al eliminarse un glicano puede deberse a que forma parte del sitio de unión para un ligando, o a que es necesario para el correcto plegado, sin ser estas posibilidades mutuamente excluyentes. Para el caso de glicoproteínas que presentan más de un glicano, hay ejemplos en los cuales algunos son necesarios para procesos de reconocimiento mientras que otros los son para el plegamiento.
Síntesis del Dolicol-Oligosacárido
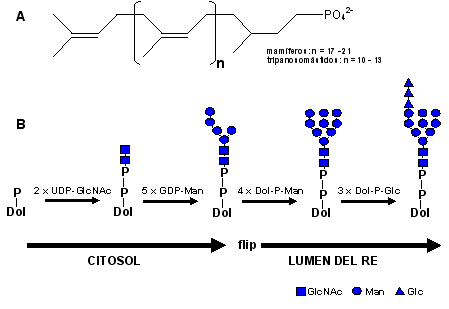
La biosíntesis de dolicol-P-P-GlNAc2Man9Glc3 ocurre sobre ambos lados de la membrana del RE (Fig. 8). El primer paso es la generación de dolicol-P-P-GlcNAc sobre el lado citosólico, el cual se sintetiza a partir de dolicol-fosfato y UDP-GlcNAc. A continuación se incorpora el segundo residuo de azúcar, el cual también proviene de UDP-GlcNAc. Los cinco residuos siguientes de manosa también se incorporan de lado citosólico, usando GDP-Man como dador. Luego el intermediario dolicol-P-P-GlcNAc2Man5 se transloca hacia el lado del lumen del RE en un proceso denominado flipping. Allí se incorporan los demás residuos de manosa y glucosa a partir de dolicol-P-manosa y dolicol-P-glucosa, respectivamente. Estos precursores son sintetizados sobre la hemicapa lumenal de la membrana a partir de dolicol-fosfato, UDP-Glc y GDP-Man. No son claras las razones para esta peculiar distribución entre las hemicapas de la membrana. Se conocen todas las enzimas de esta vía con la excepción de la encargada de la translocación del dolicol-P-P-GlcNAc2Man5, denominada "flipasa". En levaduras es posible eliminar algunos de los genes de esta vía, de forma de modificar a voluntad el oligosacárido transferido. Por ejemplo, la eliminación del gen alg5 provoca la transferencia de GlcNAc2Man9, a las glicoproteínas nacientes, aunque con menor eficiencia. Si bien en la mayor parte de los organismos la N-glicosilación se inicia con la transferencia del oligosacárido GlcNAc2Man9Glc3, en algunos se transfieren versiones más abreviadas del mismo. Por ejemplo T. cruzi, al carecer de la enzima que sintetiza dolicol-P-Glc, transfiere GlcNAc2Man9.
Figura 8. (A) Estructura del dolicol fosfato. (B) Biosíntesis del dolicol-P-P-GlcNAc2Man9Glc3.
Procesamiento de los N-Glicanos
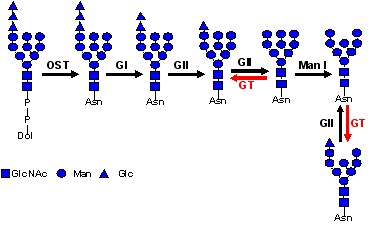
La N-glicosilación se inicia con la transferencia del mismo oligosacárido para todas las glicoproteínas. Asimismo, el procesamiento inicial que sufre en el RE es común para todas (Fig. 9). Después de la transferencia, las enzimas glucosidasa I (GI) y glucosidasa II (GII) eliminan los residuos de glucosa. Estas enzimas se encuentran en el RE, la primera unida a la membrana y la segunda soluble en el lumen. Asimismo es común que mientras la glicoproteínas está en el retículo se pierda la manosa externa de la rama B, reacción catalizada por la alfa manosidasa I del RE (Man I).
Figura 9. Procesamiento inicial de los N-glicanos en el RE.
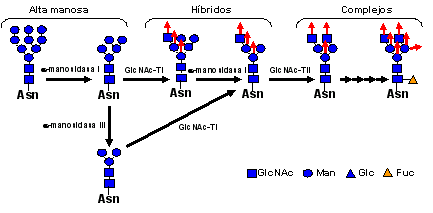
La uniformidad en este procesamiento inicial contrasta con la gran diversidad estructural de los N-glicanos presentes en las glicoproteínas maduras. Se pueden encontrar diversos tipos de N-glicanos (alta manosa, híbridos y complejos), los cuales pueden presentar otros residuos distintos de los transferidos originalmente (fucosa, ácido siálico, galactosa, xilosa, etc.). Hay que resaltar que mientras tres aminoácidos idénticos pueden formar un único péptido posible, tres hexosas iguales se pueden combinar de 176 formas diferentes. La gran diversidad de los glicanos maduros es consecuencia de la actividad de diversas glicosidasas y glicosiltransferasas localizadas principalmente en el aparato de Golgi (Fig. 10).
Figura 10. Procesamiento de los N-glicanos en el Golgi. Las flechas rojas indican sitios en donde se pueden adicionar nuevos residuos.
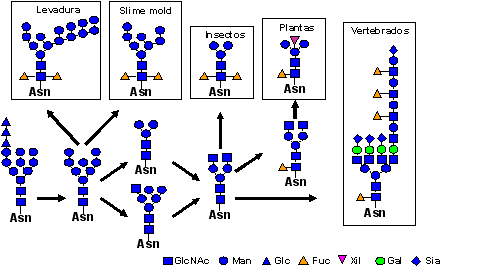
Los N-glicanos maduros preservan el "core glycan" (GlcNAc2Man3), el cual está compuesto por los cinco primeros residuos presentes en el glicano transferido inicialmente. En los distintos taxones existen vías predominantes para la maduración de los N-glicanos. Por ejemplo, en levaduras los más común es la adición de varios residuos de manosa, mientras los insectos usualmente presentan el "core glycan" modificado con fucosa, sin mostrar una mayor elongación de las ramas externas (Fig. 11).
Figura 11. Algunos de los glicanos que se encuentran frecuentemente en diferentes taxones.
Como vimos, luego de la transferencia el glicano es procesado hasta llegar al "core glycan". Sin embargo, para la síntesis de GlcNAc2Man9Glc3 se emplean una gran cantidad de precursores de alta energía. Surgen las siguientes preguntas: ¿Por qué transferir un compuesto que a continuación va a ser degradado? ¿No sería más conveniente transferir únicamente el "core glycan" y a partir de allí construir el resto del glicano? Responder estas preguntas tomó más de veinte años de trabajo. Hoy sabemos que los N-glicanos son utilizados durante la biosíntesis y plegamiento de proteínas en el RE como un sistema que codifica información acerca de su estado conformacional. Puesto que durante su plegamiento las proteínas deben enfrentar problemas similares, independientemente de su función biológica, las células aprovechan un sistema común basado N-glicanos para lograrlo.
La reglucosilación de proteínas
Una observación inicial que sugirió funciones tempranas de la N-glicosilación fue la detección en T. cruzi del oligosacárido GlcNAc2Man9Glc (21). En la mayor parte de los organismos la presencia de este compuesto se explica por la deglucosilación parcial del oligosacárido trasferido, sin embargo como T. cruzi transfiere GlcNAc2Man9, la presencia de glicanos monoglucosilados sólo puede explicarse por una adición de glucosa posterior a la glicosilación inicial (22). Este residuo de glucosa está presente mientras las glicoproteínas se encuentran en el RE, y generalmente no se lo detecta una vez que arribaron a su destino final. Este hallazgo incentivó la búsqueda de esta reacción en otros organismos, encontrándose conservada en prácticamente todos los eucariontes con la excepción de S. cerevisiae. La glucosa que se agrega es la última que fue removida por la GII. Asimismo, esta reacción puede darse sobre glicanos que han perdido una o dos manosas, aunque con menor eficiencia. Por lo tanto en el esquema del procesamiento inicial de los N-glicanos debemos incluir esta nueva reacción (Fig. 12).
Figura 12. Procesamiento de los N-glicanos en el Golgi.
La enzima responsable de la reglucosilación se denomina UPD-Glc:glicoproteína glucosiltransferasa (GT o UGGT). Esta enzima utiliza como aceptor a una glicoproteína conteniendo oligosacáridos de alta manosa (GlcNAc2Man9, GlcNAc2Man8 y GlcNAc2Man7) y como dador de glucosa al UDP-Glc. En su extremo C-terminal presenta una secuencia KDEL de retención, siendo la única glicosiltransferasa soluble en el lumen del RE (23). Los productos de la reacción catalizada por la GT son la proteína glucosilada y UDP. En los primeros intentos para purificar esta enzima se probó como sustrato aceptor a la tiroglobulina bovina y como dador al UDP-[14C]-Glc. En teoría, luego de incubar los microsomas con esta mezcla y de precipitar la tiroglobulina con ácido tricloroacético se podría medir la cantidad de glucosa incorporada mediante la radioactividad asociada al precipitado. Si bien en estas condiciones es posible medir la reacción, la actividad enzimática que se obtiene es bastante pobre. Por este motivo la purificación de la GT estuvo frenada durante varios años. Eventualmente se pensó que la tiroglobulina no era un buen aceptor porque sus N-glicanos podrían estar ocultos, siendo inaccesibles a la enzima. Por este motivo se probó desnaturalizarla previamente con urea y calor. Al usar este nuevo sustrato se encontró que la reacción era unos dos órdenes de magnitud más rápida que la medida con el aceptor en condiciones nativas. Este nuevo ensayo sirvió como base para purificar la GT, y luego de secuenciar su extremo amino terminal se la pudo clonar. Si la teoría de la baja exposición de los glicanos en la tiroglobulina nativa fuera correcta, un oligosacárido de alta manosa debería se un aceptor óptimo. Contrariamente a lo esperado, estos oligosacáridos son aceptores bastante pobres. Estas observaciones muestran que la GT reconoce los glicanos de alta manosa que están unidos a proteínas desnaturalizadas, mientras que cuando las mismas se encuentran en forma nativa son sustratos pobres. Una observación similar se realizó con diversas glicoproteínas, de forma que la GT reconocería algún aspecto común a las glicoproteínas desplegadas (¿Mayor movilidad de la cadena peptídica? ¿Exposición de residuos hidrofóbicos?).
La serendipia en el descubrimiento científico
Serendipia (del inglés serendipity): hallazgo científico afortunado e inesperado que se realiza de forma casual. Fue acuñado por el escritor inglés Horace Walpole en 1754 a partir de un cuento persa del siglo XVIII llamado «Los tres príncipes de Serendip», en donde los protagonistas, unos príncipes de la isla Serendip (nombre árabe para Sri Lanka), resolvía sus problemas a través de increíbles casualidades.
El descubrimiento de la reglucosilación ocurrió gracias a dos hechos fortuitos, el sistema biológico utilizado (T. cruzi) y la forma de marcación ([14C]-Glc). Solamente en un kinetoplástido es posible detectar esta reacción, porque estos organismos transfieren glicanos desprovistos de residuos de glucosa. Sin embargo con esto no alcanza. Usualmente para marcar oligosacáridos se emplea [3H]-Man, dado que por su mayor radioactividad específica es más barato que el [14C]-Glc. De haberse utilizado manosa tritiada hubiera sido imposible detectar los derivados monoglucosilados. Esto se debe a que el análisis de glicanos se realiza mediante cromatografía en papel, en donde los oligosacáridos se detectan por su radioactividad (también se puede usar HPLC). En las marcaciones con [3H]-Man los picos principales que se detectan son GlcNAc2Man9, GlcNAc2Man8 y GlcNAc2Man7, mientras que sus derivados monoglucosilados son de muy difícil detección. En cambio, cuando se marca con [14C]-Glc, aunque eventualmente todos los azúcares terminan marcados, en tiempos cortos de incubación lo primero que se marca son los oligosacáridos monoglucosilados, permitiendo así su detección. Si bien la marcación con [14C]-Glc es bastante más cara, gracias a la ineficiencia de la Aduana a mediados de los años 70 no era conveniente marcar con [3H]-Man. Resulta que la alta actividad específica de este compuesto hace que sufra un proceso de auto degradación denominada radiólisis y por ello no es posible almacenarlo por mucho tiempo. Este problema no se da con el [14C]-Glc. Cuando llegaban los envíos del exterior era frecuente que fueran retenidos en la Aduana durante meses por trabas burocráticas de lo más variadas, de forma que cuando la [3H]-Man finalmente llegaba al laboratorio era inservible. Este es un buen ejemplo en donde la ineficiencia burocrática adelantó el progreso de la ciencia (no conozco ningún otro).
Control de calidad de plegamiento
En paralelo al descubrimiento de la GT se purificó una proteína con actividad de lectina por oligosacáridos monoglucosilados. Inicialmente esta lectina fue aislada por su capacidad para unir calcio, y dada su localización en el RE se la denominó calreticulina (CRT) (24, 25). Al poco tiempo se aisló una proteína paráloga a CRT, la cual está anclada a la membrana del RE y que fue bautizada como calnexina (CNX). Asimismo, desde hacía tiempo se conocía la GII, enzima que revierte la actividad de GT, pero que a diferencia de GT su actividad no es afectada por el estado conformacional de sus sustratos. Por lo tanto a principios de los años 90 se conocían los siguientes componentes:
· CRT y CNX: lectinas residentes del RE que unen glicanos monoglucosilados de alta manosa
· GT: enzima que glucosila glicoproteínas de alta manosa en conformaciones no nativas.
· GII: enzima que hidroliza la glucosa de glicoproteínas monoglucosiladas independientemente de su estado conformacional.
En el año 1994 fue publicado un trabajo que cambió la visión del plegamiento de proteínas en el RE (26). En este artículo se combinaron estos componentes en un mecanismo denominado control de calidad de plegamiento de glicoproteínas (QC). En este ciclo las proteínas ingresan al RE, son glicosiladas con GlcNAc2Man9Glc3 y a continuación se remueven las dos glucosas terminales, generando GlcNAc2Man9Glc (Fig. 13). En esta etapa la glicoproteínas son unidas por CRT y/o CNX, reteniéndolas en el RE. Eventualmente el último residuo de Glc es removido por la GII, y las glicoproteínas se disocian de las lectinas. En esta instancia, si la glicoproteína adquirió se estructura nativa podrá seguir su camino por la vía secretoria. Por el contrario, de no haberse plegado correctamente será reconocida por la GT, quien vuelve a añadir el último residuo de glucosa y regenera el complejo con las lectinas. De esta forma las proteínas que no terminaron de plegarse son retenidas en el RE. Prácticamente todas las glicoproteínas son sometidas a este control de calidad, el cual aumenta la eficiencia del plegamiento al otorgarle a las proteínas más oportunidades para plegarse correctamente. En general las chaperonas se unen a proteínas desplegadas en un ciclo acoplado a la hidrólisis de ATP. En el fondo el ciclo de CRT/CNX hace algo similar, en donde el papel del ATP es reemplazado por UDP-Glc. Este ciclo puede visualizarse como una chaperona clásica en donde las funciones de reconocimiento de defectos estructurales (GT), unión de proteínas mal plegadas (CRT/CNX) e hidrólisis de UDP-Glc (GT y GII) fueron distribuidos entre varias proteínas. El QC no sólo supervisa la adquisición de la estructura terciaria, sino que también opera durante el ensamblado de oligómeros. Se ha demostrado, por ejemplo para el receptor de linfocitos T, que las subunidades del oligómero son retenidas en el RE hasta que se completa su ensamblado (27). Y de hecho se ha visto en experimentos libres de células que la GT puede reconocer subunidades "huérfanas" (ver abajo).
Como todo control de calidad, este sistema puede presentar dos posibles fallas: por defecto y por exceso. En el primer caso, proteínas que presentan errores en su conformación evaden el QC. Esto puede tener consecuencias desastrosas porque estas proteínas pueden generar agregados tóxicos. Por el contrario, un exceso de celo del sistema podría retener en el RE proteínas biológicamente activas. Existen ejemplos para ambos casos. El caso de la fibrosis quística es muy ilustrativo. Esta enfermedad se origina por la ausencia (o deficiencia) de una proteína de la membrana plasmática que funciona como canal de cloruro denominada CFTR, siendo la enfermedad genética de mayor incidencia en caucásicos (28). Las personas afectadas tienen mutaciones en el gen de CTR que imposibilitan su salida del RE, siendo la más común la ausencia de un residuo de fenilalanina en la posición 508 (DF508). Estas proteínas mutantes poseerían actividad biológica si arribaran a la membrana plasmática, sin embargo nunca llegan a su destino final porque son retenidas en el RE por el QC. De hecho la variante silvestre del CFTR presenta una eficiencia de maduración muy baja, y sólo el 10 % de la proteína sintetizada llega a su destino final. Para tratar este tipo de patologías se está explorando la posibilidad de modular al sistema de QC de forma de hacerlo menos eficiente para algunos sustratos (por ejemplo estabilizando el CFTR). En el fondo estos problemas surgen porque unos de los principales dilemas del QC es distinguir las proteínas que están en proceso de plegarse correctamente de aquellas que presentan fallas irreversibles, siendo necesario un delicado equilibrio entre la maquinaria de plegado y la encargada de degradar proteínas.
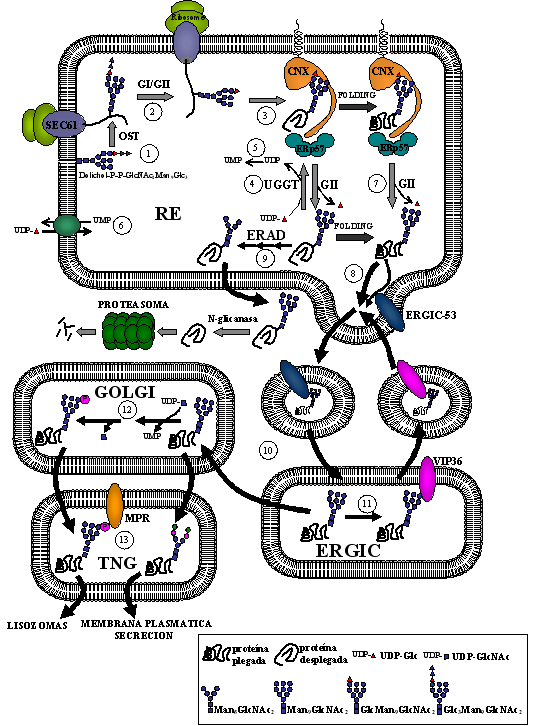
Figura 13. Rol de los N-glicanos en la vía secretoria. (1) Transferencia inicial de N-glicano. (2) Corte de las dos glucosas externas por GI y GII. (3) Unión de los intermediarios monoglucosilados a CRT/CNX. (4 y 5) Reglucosilación por la GT de las proteínas mal plegadas y generación de UDP. (6) Ingreso de UDP-Glc al lumen del RE. (7) Hidrólisis de la última glucosa por la GII. (8) Exportación de proteínas del RE. (9) Degradación de proteínas mal plegadas por el ERAD. (10) Exportación de glicoproteínas hacia el ERGIC por ERGIC-53. (11) Retorno de glicoproteínas desde el ERGIC por VIP36. (12) Generación de Man-6-P en el Golgi. (13) Redireccionamiento de proteínas hacia el lisosoma.
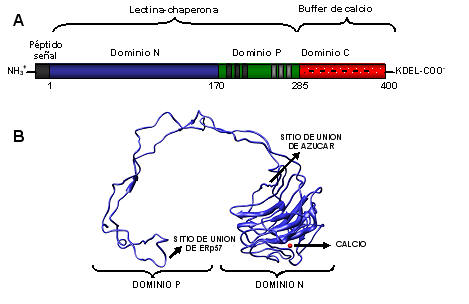
Estructura de CRT y CNX
CRT y CNX tienen una identidad de secuencia cercana al 40 % y presentan una estructura muy similar. La principal diferencia es la presencia en CNX de un segmento hidrofóbico cerca del extremo carboxilo terminal que la ancla a la membrana del RE. Ambas proteínas presentan un dominio globular sobre el extremo amino terminal formado por dos hojas beta antiparalelas (dominio N). En este dominio, que es muy similar a las lectinas de plantas leguminosas, reside el sitio de unión para oligosacáridos monoglucosilados. Una característica única de estas lectinas es la presencia de un dominio alargado protruyente, muy rico en residuos de prolina (dominio P). Si bien la actividad de lectina no depende de la presencia del dominio P, se ha demostrado que mejora su actividad como chaperona. A partir de experimentos de difracción de rayos X se resolvió la estructura de la zona lumenal de CNX (dominio P más dominio N), y mediante resonancia magnética nuclear conocemos la estructura del dominio P de CRT (Fig. 14) (29). También contamos con fotografías de microscopía electrónica de CRT en donde se observa un dominio globular (dominio N) unido a una zona elongada de gran flexibilidad (dominio P) (30). No disponemos de la estructura del complejo entre las lectinas y sus ligandos, aunque la estructura cristalográfica de CNX sugiere posible un mecanismo de unión.
Figura 14. (A) Distribución de dominios de CRT. (B) Estructura del dominio lumenal de CNX (código PDB: 1JHN).
La gran flexibilidad del dominio P en principio podría haber evitado la obtención de cristales con la calidad necesaria para la difracción de rayos X. Sin embargo la estructura de esta proteína muestra el dominio P en una conformación definida. Esto se comprende al observar las proteínas vecinas en el cristal, en donde el dominio P rodea al dominio N de una proteína contigua (Fig. 15).
Figura 15. Estructura del dominio lumenal de CNX en donde se muestra la proteína más cercana la celda cristalina (PDB: 1JHN).
Posiblemente mientras una glicoproteína se une al dominio N a través de su glicano, el dominio P "abrazaría" a su porción peptídica. Este complejo protegería al sustrato de interacciones no deseadas, previniendo su agregación. Si bien no sabemos qué grado de desplegado presenta un sustrato cuando se une a CRT o CNX, evidencia obtenida in vivo sugiere que se unen a intermediarios avanzados de plegamiento, posiblemente como consecuencia de la mayor actividad de GT para con este tipo de estructuras (ver abajo). Por otra parte, la punta del dominio P interacciona con ERp57, una PDI que presenta cierta especificidad por glicoproteínas. De esta forma mientras la glicoproteína se mantiene unida a CRT/CNX, ERp57 ayuda en la formación de los puentes disulfuro nativos. Una comparación de la distribución de dominios entre la PDI clásica y ERp57 es reveladora. Ambas proteínas poseen cuatro dominios homólogos a tioredoxina (a-b-b´-a´), de los cuales sólo los dominios a y a´ presentan el sitio activo para la isomerización. En la PDI los dominios b y b´ están dedicados a unir al sustrato, mientras que en ERp57 estos dominios se unen a la punta del dominio P. De esta forma la presentación de los sustratos a ERp57 estaría a cargo de CRT y CNX, explicando su mayor selectividad por glicoproteínas.
Reconocimiento por GT
De los cuatro componentes del QC, el único sensible al estado conformacional de los sustratos es la GT. Esta proteína está presente en prácticamente todas las células eucariontes (con la notable excepción de S. cerevisiae) y su tamaño ronda los 160-170 kDa. Está conformada como mínimo por dos dominios. El dominio carboxilo terminal está conservado, comprende cerca del 20 % de la secuencia, une UDP-Glc y allí se localiza el sitio activo de la enzima. El dominio N terminal abarca el resto de la proteína, presenta una gran variedad de secuencia y se supone que es el encargado de reconocer al polipéptido mal plegado. Proteínas quiméricas construidas con dominios N y C de especies diferentes poseen actividad biológica. Llamativamente en algunas especies existe un segundo gen con alta homología con GT. En humanos los genes para GT se llaman huggt1 y huggt2, y si bien ambos se expresan sólo el primero posee actividad biológica.
Experimentos preliminares revelaron que la GT presenta una preferencia por proteínas desplegadas (31). Recientemente se pudo demostrar que dentro de la miríada de las posibles conformaciones que puede adoptar una proteína mal plegada, la GT prefiere a aquellas que son más compactas y que exponen parches hidrofóbicos. Esto se vio utilizando una familia de proteínas derivadas del inhibidor de quimiotripsina 2 que presenta cortes progresivos en su extremo C-terminal (32, 33). La capacidad de esta familia como sustrato de GT es muy similar a su habilidad para unir el ácido anilino naftalensulfónico (ANS). Este compuesto fluorescente se une a parches hidrofóbicos expuestos, siendo muy útil para detectar intermediarios colapsados de plegamiento. Asimismo, usando como sustrato la aglutinina de soja (SBA) se pudo demostrar que la GT puede reconocer oligómeros desensamblados (34). La SBA es un homotetrámero que puede desensamblarse en monómeros al incubarse con concentraciones bajas de urea. Al disociar la SBA la actividad de GT aumenta de forma paralela a la fracción molar de monómero. De forma similar a lo observado con el CI2, existe aquí una excelente correlación entre unión de ANS y reconocimiento por GT. En este caso el ANS se une a un parche hidrofóbico que se encuentra oculto en la superficie de contacto entre las subunidades del tetrámero, el cual se expone cuando se lo disocia. Estos experimentos fueron hechos en sistemas libres de células y ambos sugieren que la GT participaría durante las etapas avanzadas del plegamiento. Esto se ha podido confirmar en células de T. cruzi siguiendo como sustrato modelo la cruzipaína, una abundante proteasa lisosomal (35). Al estudiarse el estado de plegamiento de la cruzipaína se ve que cuando se encuentra asociada a CRT tiene formados gran parte de sus puentes disulfuro. Como en este sistema biológico la única forma que tienen las glicoproteínas de asociarse a CRT es a través de la actividad de GT, estas observaciones sugieren que la enzima reconoció a la cruzipaína en etapas avanzadas de su maduración conformacional. No existe información estructural de la GT, y por lo tanto desconocemos cómo hace para reconocer a sus sustratos.
Los N-Glicanos más allá del RE
El movimiento de glicoproteínas entre el RE y ERGIC (compartimiento intermedio entre el RE y el Golgi) está mediado en varios casos por receptores con actividad de lectina. Existen al menos dos lectinas que ciclan permanentemente entre ambos compartimentos, ERGIC-53 y VIP36. Estas lectinas presentan un corto segmento del lado citosólico con la secuencia KKFF (ERGIC-53) y KRFY (VIP36) responsable de este movimiento (36). Ambas lectinas tienen afinidad por glicanos de alta manosa. ERGIC-53 participa en la salida del RE de los factores de coagulación V y VIII, y de hecho ciertos tipos de hemofilia se deben a mutaciones en esta lectina. Asimismo la salida de algunas proteasas lisosomales (catepsinas Z y C) también está mediada por ERGIC-53. La actividad de lectina de ERGIC-53 es dependiente del calcio y es óptima a pH 7.4, las condiciones del lumen del RE. Por este motivo la unión de los ligandos a ERGIC-53 se ve favorecida en el RE. Cuando el complejo llega al ERGIC encuentra un pH ligeramente menor y con una menor concentración de calcio, lo cual gatilla su disociación. La dependencia con el pH de la actividad de VIP36 inversa, y se presume que su función sería llevar de vuelta a algunas glicoproteínas al ER, posiblemente a aquellas que aún no finalizaron su plegamiento y que escaparon del QC (37). No se sabe si VIP36 presenta mayor afinidad por glicoproteínas mal plegadas. En el aparato de GOLGI existe una bifurcación en la vía secretoria, en donde se dividen las proteínas que van a los lisosomas de aquellas que terminan en la membrana plasmática o son secretadas. Aquí también los glicanos cumplen un papel fundamental. Las manosas de las glicoproteínas lisosomales son modificadas con un fosfato en la posición 6, el cual sirve como señal de reconocimiento para receptores específicos (MPR) que las conduce a los lisosomas. Estos sistemas muestran que los glicanos son utilizados en varias instancias decisivas a lo largo de la vía secretoria.
Perspectivas
Existen muchas preguntas abiertas acerca de las funciones biológicas de los glicanos. No comprendemos el papel que cumplen durante el reconocimiento de las proteínas que van a ser sustratos del ERAD. Si bien la pérdida de manosas cumple un rol fundamental, no sabemos cómo el sistema distingue entre las proteínas que están en un camino productivo de plegamiento de aquellas irreversiblemente mal plegadas. Aparentemente la baja actividad de la manosidasa del RE les daría tiempo a las glicoproteínas para plegarse, funcionando como una especie de temporizador. Por otra parte, las glicoproteínas sustratos del ERAD son demanosiladas hasta GlcNAc2Man5, y recién entonces pueden ser retrotranslocadas al citosol. En principio debería existir alguna flipasa que permita el pasaje del glicano a través de la membrana, y dado que en la vía biosintética del dolicol-P-P-oligosacárido el paso de "flipping" sucede cuando el glicano intermediario presenta la misma estructura es tentador pensar que la misma proteína se encargaría de ambos procesos. Por otro lado, recién ahora comenzamos a comprender la compleja regulación de la actividad de la oligosacariltransferasa. Variaciones en los patrones de expresión de las subunidades catalíticas de este complejo (STT3A y STT3B) podría afectar el nivel de glicosilación de ciertas hormonas proteicas, añadiendo un nuevo nivel de regulación de su actividad biológica. Por último, si bien la glicosilación de proteínas hasta hace poco se suponía circunscripta a la vía secretoria, hoy sabemos que muchas proteínas citosólicas pueden ser O-glicosiladas por GlcNAc, modulando diversos procesos de señalización celular. En definitiva, la glicosilación de proteínas pasó de ser una curiosidad bioquímica a jugar un papel clave en diversos procesos de reconocimiento celular y maduración conformacional de las glicoproteínas.
Bibliografía
1. Osborne AR, Rapoport TA, y van den Berg B, (2005) Protein translocation by the Sec61/SecY channel. Annu. Rev. Cell. Dev. Biol. 21:529-50.
2. Kleizen B, Braakman I, (2004) Protein folding and quality control in the endoplasmic reticulum. Curr. Opin. Cell. Biol. 16:343-9.
3. Paulsson K, Wang P, (2003) Chaperones and folding of MHC class I molecules in the endoplasmic reticulum. Biochim. Biophys. Acta. 1641:1-12.
4. Gething MJ, (1999) Role and regulation of the ER chaperone BiP. Semin. Cell. Dev. Biol. 10:465-72.
5. Genevaux P, Georgopoulos C, Kelley WL, (2007) The Hsp70 chaperone machines of Escherichia coli: a paradigm for the repartition of chaperone functions. Mol. Microbiol. 66:840-57.
6. Ni M, Lee AS, (2007) ER chaperones in mammalian development and human diseases. FEBS Lett. 581:3641-51.
7. Vembar SS, Brodsky JL, (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell. Biol. 9:944-57.
8. Malhotra JD, Kaufman RJ, (2007) The endoplasmic reticulum and the unfolded protein response. Semin. Cell. Dev. Biol. 18:716-31.
9. Sommer T, Jarosch E, (2002) BiP binding keeps ATF6 at bay. Dev Cell. 3:1-2.
10. Gass JN, Jiang HY, Wek RC, Brewer JW, (2008) The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol. Immunol. 45:1035-43.
11. Riemer J, Bulleid N, Herrmann JM, (2009) Disulfide formation in the ER and mitochondria: two solutions to a common process. Science 324:1284-7.
12. Appenzeller-Herzog C, Ellgaard L, (2008) The human PDI family: versatility packed into a single fold. Biochim. Biophys. Acta 1783:535-48.
13. Jessop CE, Tavender TJ, Watkins RH, Chambers JE, Bulleid NJ, (2009) Substrate specificity of the oxidoreductase ERp57 is determined primarily by its interaction with calnexin and calreticulin. J. Biol. Chem. 284:2194-202.
14. Kelleher DJ, Gilmore R, (2006) An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16:47R-62R.
15. Apweiler R, Hermjakob H, Sharon N, (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta. 1473:4-8.
16. Castro O, Movsichoff F, Parodi AJ, (2006) Preferential transfer of the complete glycan is determined by the oligosaccharyltransferase complex and not by the catalytic subunit. Proc. Natl .Acad. Sci. U S A. 103:14756-60.
17. Ruiz-Canada C, Kelleher DJ, Gilmore R, (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 23:272-83.
18. Weerapana E, Imperiali B, (2006) Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology 16:91R-101R.
19. Solá RJ, Rodríguez-Martínez JA, Griebenow K, (2007) Modulation of protein biophysical properties by chemical glycosylation: biochemical insights and biomedical implications. Cell. Mol. Life Sci. 64:2133-52.
20. O'Connor SE, Pohlmann J, Imperiali B, Saskiawan I, Yamamoto K, (2001) Probing the effect of the outer saccharide residues of N-linked glycans on peptide conformation. J. Am. Chem. Soc. 123:6187-8.
21. Trombetta SE, Bosch M, Parodi AJ, (1989) Glucosylation of glycoproteins by mammalian, plant, fungal, and trypanosomatid protozoa microsomal membranes. Biochemistry 28:8108-16.
22. Bosch M, Trombetta S, Parodi AJ, (1988) Synthesis of dolichol derivatives and protein glycosylation in trypanosomatids. Biochem. Soc. Trans. 16:268-71.
23. Trombetta SE, Parodi AJ, (1992) Purification to apparent homogeneity and partial characterization of rat liver UDP-glucose:glycoprotein glucosyltransferase. J. Biol. Chem. 267:9236-40.
24. Waisman, DM, Salimath, BP, Anderson, MJ, (1985) Isolation and characterization of CAB-63, a novel calcium-binding protein, J. Biol. Chem. 260: 1652-60
25. Fliegel L, Burns K, MacLennan DH, Reithmeier RA, Michalak M, (1989) Molecular cloning of the high affinity calcium-binding protein (calreticulin) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 264:21522-8.
26. Hammond C, Braakman I, Helenius A, (1994) Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control.
Proc. Natl. Acad. Sci. U S A. 91:913-7.
27. Gardner TG, Kearse KP, (1999) Modification of the T cell antigen receptor (TCR) complex by UDP-glucose:glycoprotein glucosyltransferase. TCR folding is finalized convergent with formation of alpha beta delta epsilon gamma epsilon complexes. J. Biol. Chem. 274:14094-9.
28. Hirsch C, Jarosch E, Sommer T, Wolf DH, (2004) Endoplasmic reticulum-associated protein degradation--one model fits all? Biochim. Biophys. Acta 1695:215-23.
29. Schrag JD, Bergeron JJ, Li Y, Borisova S, Hahn M, Thomas DY, Cygler M, (2001) The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol. Cell. 8:633-44.
30. Tan Y, Chen M, Li Z, Mabuchi K, Bouvier M, (2006) The calcium- and zinc-responsive regions of calreticulin reside strictly in the N-/C-domain. Biochim. Biophys. Acta 1760:745-53.
31. Sousa M, Parodi AJ, (1995) The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 14:4196-203.
32. Caramelo JJ, Castro OA, de Prat-Gay G, Parodi AJ, (2004) The endoplasmic reticulum glucosyltransferase recognizes nearly native glycoprotein folding intermediates. J. Biol. Chem. 279:46280-5.
33. Caramelo JJ, Castro OA, Alonso LG, de Prat-Gay G, Parodi AJ, (2003) UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc. Natl. Acad. Sci. U S A 100:86-91.
34. Keith N, Parodi AJ, Caramelo JJ, (2005) Glycoprotein tertiary and quaternary structures are monitored by the same quality control mechanism. J. Biol. Chem. 280:18138-41.
35. Labriola C, Cazzulo JJ, Parodi AJ, (1995) Retention of glucose units added by the UDP-GLC:glycoprotein glucosyltransferase delays exit of glycoproteins from the endoplasmic reticulum. J. Cell. Biol. 130:771-9.
36. Hauri HP, Nufer O, Breuza L, Tekaya HB, Liang L, (2002) Lectins and protein traffic early in the secretory pathway. Biochem. Soc. Symp. 69:73-82.
37. Kamiya Y, Kamiya D, Yamamoto K, Nyfeler B, Hauri HP, Kato K, (2008) Molecular basis of sugar recognition by the human L-type lectins ERGIC-53, VIPL, and VIP36. J. Biol. Chem. 283:1857-61.
ISSN 1666-7948
www.quimicaviva.qb.fcen.uba.arRevista QuímicaViva
Número 2, año 8, Agosto 2009
quimicaviva@qb.fcen.uba.ar