La bioinformática estructural o la realidad virtual de los medicamentos.
Marcelo A. Martí and Adrian A. Turjanski
Departamento de Química Inorgánica, Analítica y Química Física e INQUIMAE, y Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales
Universidad de Buenos Aires.
Ciudad Universitaria, Pab. II, 4o piso, 1428 Buenos Aires. Argentina. Tel/fax: 5411-4576-3342
Recibido el 26/03/09. Aceptado el 10/04/09.
Resumen
La bioinformática estructural consiste, entre otras cosas, en realizar una simulación del comportamiento de biomoléculas, principalmente proteínas y sus entornos, en diferentes situaciones. Al igual que un simulador de vuelo nos puede decir si un piloto se encuentra en condiciones de conducir con éxito un avión a su destino, la simulación con las biomoléculas puede establecer la capacidad de una droga para inhibir algunas enzimas o la posibilidad que dos proteínas puedan interactuar entre ellas. De este modo la bio informática contribuirá a un cambio en el paradigma del diseño de drogas permitiendo una mayor rapidez en su descubrimiento y en el proceso de optimización.
Hay ejemplos exitosos de este nuevo paradigma como el caso de los inhibidores selectivos COX-2 como el Celecoxib usados en enfermedades inflamatorias crónicas, los inhibidores de la enzima convertidora de angiotensina (ECA) como el Captopril, empleados en el tratamiento de la hipertensión y diversos compuestos para el tratamiento del HIV.
Palabras claves:
bioinformática estructural – nuevo paradigma de desarrollo de medicamentos – diseño racional de fármacos – desarrollo de fármacos in silico
Structural bioinformatics: the drugs virtual reality game
Abstract
Structural Bioinformatics consist in simulating biomolecules, mainly proteins and their environment in different situations. And just like a flight simulator can tell us, if a pilot is able to bring the plane safely to destiny, biomolecular simulations can determine the ability of a drug to inhibit certain enzyme, or the possibility of two proteins to interact with each other. In this way structural bioinformatics contributed to a change in the drug development paradigm allowing a faster drug discovery and optimization process. Successful examples of the new paradigm are COX-2 selective inhibitors used in chronic inflammations, ACE inhibitor captopril and several compounds to treat HIV among others.
Key words:
structural bioinformatics - new paradigm in drug development – rational drug design – in ilico drug development
Introducción
En la transición entre el siglo XX y XXI comenzaron a aparecer en los salones comerciales de videojuegos, un nuevo tipo de juegos que permiten al usuario sentirse manejando un auto a alta velocidad o participando de una batalla épica entre otras cosas. Estos juegos que se denominan de realidad virtual, “simulan” entornos sintéticos que el sujeto percibe como reales, pero que solo existen dentro del ordenador. De manera análoga, la bioinformática estructural consiste, entre otras cosas, en realizar una simulación de biomoléculas, principalmente proteínas y sus entornos, en diferentes situaciones. Y así como un simulador de vuelo es capaz de determinar si un piloto es capaz de llevar el avión a destino, la bioinformática estructural puede determinar si cierta droga es capaz de inhibir una enzima o si un par de proteínas pueden interactuar entre sí. De esta manera la bioinformática estructural ha contribuido a un cambio de paradigma en el desarrollo de medicamentos contribuyendo al diseño racional y acelerando los procesos de identificación y mejoramiento de nuevas drogas. Ejemplos exitosos de este nuevo paradigma son: los inhibidores COX-2 selectivos como el Celecoxib usados en enfermedades inflamatorias crónicas, los inhibidores de la enzima convertidora de angiotensina (ECA) como el Captopril, empleados en el tratamiento de la hipertensión y diversos compuestos para el tratamiento del HIV.
¿Que es la bioinformática estructural?
Definir la bioinformática, al igual que otras jóvenes ramas de la ciencia de carácter interdisciplinario, no es tarea fácil, ya que los límites son difusos. Algunos la definen únicamente como el desarrollo de bases de datos para la manipulación de la información genómica y estructural. Otros en cambio, consideran que abarca toda la biología computacional. El dogma central de la biologίa molecular establece que el ADN es transcripto a ARN mensajero y que éste es traducido luego en una proteína, que al adquirir su estructura tridimensional, cumple su rol en el funcionamiento del organismo. La bioinformática estudia el flujo de información en todos los estadίos del dogma central, como la organización y la regulación de los genes en la secuencia del ADN, la identificación de las zonas de transcripción del ADN, la predicción de la estructura de las proteínas a partir de su secuencia y el análisis de la función molecular de las biomacromoléculas.
El gran número de estructuras 3D de biomacromoléculas que se han resuelto en las últimas décadas y su deposición en bases de datos como el “Protein Data Bank” (www.pdb.org) han generado el desarrollo de una subdisciplina dentro de la bioinformática: La bioinformática estructural. La bioinformática estructural es entonces la disciplina de la ciencia que se dedica al análisis, caracterización y visualización de estructuras biomacromoleculares, principalmente proteínas, ADN y ARN, y sus interacciones mediante técnicas computacionales. Al igual que otras disciplinas la bioinformática estructural tiene dos objetivos generales: La creación de métodos computacionales para manipular, ordenar y analizar la información generada mediante experimentos “húmedos”** y la aplicación de estos métodos para resolver problemas de índole biológico y así generar nuevo conocimiento. Claramente estos objetivos están interrelacionados, dado que la validación de las metodologías involucra su uso exitoso en la resolución de problemas específicos.
**lo opuesto a un experimento seco ("in sílico")
La bioinformática ha contribuido enormemente al boom de la biologίa molecular mediante el desarrollo de algoritmos para el análisis, clasificación y determinación de secuencias de ADN y proteínas, como los algoritmos de alineamiento de secuencias de a pares. La bioinformática estructural no se ha quedado atrás y ha tenido un aporte significativo en el desarrollo de la biología estructural. Por ejemplo, mediante el conocimiento de las características fisicoquímicas de los componentes de las biomacromoléculas, (aminoácidos y ácidos nucleicos) se han desarrollado algoritmos que permiten generar modelos estructurales a partir de información obtenida por técnicas de Rayos X y RMN (Resonancia Magnética Nuclear). En el comienzo del siglo XXI, el extraordinario avance en la secuenciación de genomas, expresión de proteínas y la cristalografía a gran escala cambia radicalmente el panorama de la biología, y abre una plétora de nuevas oportunidades para el uso de las estructuras tridimensionales de proteínas en la búsqueda de fármacos. La bioinformática estructural tiene entonces un rol primordial; usar la abundante información estructural para identificar, diseñar y optimizar nuevas drogas, que sean específicas y potentes.
Bases de la Bioinformática Estructural
Para comprender las estructuras de las biomacromoléculas y sus posibles interacciones se deben conocer las bases fisicoquímicas que determinan las interacciones moleculares de manera tal de generar modelos que permitan simular in-silico (hecho por computadora o simulación por computadora) lo que ocurre in-vivo. La mecánica cuántica, que surgió a principios del siglo XX, permite explicar el comportamiento de la materia en general, pero es en el nanomundo de las moléculas donde sus resultados divergen sustancialmente de los obtenidos con la mecánica clásica. El desarrollo de computadoras cada vez más eficientes durante las últimas décadas llevó al desarrollo de algoritmos computacionales basados en la mecánica cuántica que permiten el estudio detallado de las propiedades de pequeñas moléculas. Sin embargo, dado el alto costo computacional de estos algoritmos su aplicación se limita a moléculas formadas por no más de 100 átomos y a unas pocas conformaciones. A medida que el tamaño de la molécula crece, como en el caso de las proteínas que van desde unos pocos miles a cientos de miles de átomos, crece vertiginosamente el número de conformaciones que estas pueden adoptar. En este sentido, al estudiar la interacción de una droga con una proteína se deben evaluar la ubicación y orientación de la droga a lo largo de la superficie de la proteína teniendo en cuenta los posibles cambios conformacionales de la droga y de la proteína.
Para poder trabajar con las biomacromoléculas de manera eficiente se fueron desarrollando diferentes potenciales, denominados clásicos, que modelan las interacciones de manera simplificada pero que se ajustan bien a los resultados experimentales y a lo esperado por la mecánica cuántica. La idea general es usar un algoritmo que permita el muestreo de diferentes conformaciones del sistema en estudio y calcular la “energía” de las conformaciones sometidas al muestreo mediante los potenciales simplificados. Estos potenciales se pueden dividir en dos tipos:
1) Los llamados de primeros principios, refiriéndose a que son potenciales transferibles basados en las características fisicoquímicas de los componentes y que no usan información biológica del caso particular en estudio. Un ejemplo de potencial de primeros principios ampliamente utilizado es el del programa AMBER. El mismo, simula las interacciones entre las densidades electrónicas de átomos que no están unidos covalentemente, mediante un potencial coulombico que describe la interacción electrostática y un potencial del tipo Lennard-Jones para describir las fuerzas denominadas de Van der Waals. En la bibliografía se han generado cientos de estos potenciales que varían en el detalle con el que describen las interacciones, pudiendo tener desde decenas de parámetros para cada átomo de la proteína o potenciales que reducen el problema sólo con unos pocos parámetros por proteína. Los potenciales más detallados, llamados de all-atom (todos los átomos), tienen una gran aplicabilidad para predecir cambios conformacionales pequeños en la estructura de proteínas, evaluar efectos de mutaciones y predecir la interacción droga-proteína o proteína-proteína. Los potenciales menos detallados, reciben el nombre de coarse-grain (grano grueso) y se usan en general cuando los cambios conformacionales son más pronunciados, como al estudiar el mecanismo de plegamiento de una proteína, o cuando se trabaja con proteínas de alto peso molecular. El costo computacional de un potencial all-atom es muy elevado.
2) En el otro tipo de potenciales, que se pueden denominar estadísticos, los parámetros usados para calcular la energía son generados a partir de la vasta información estructural sobre las proteínas depositada en las bases de datos y que se encuentra disponible. Por ejemplo, al estudiar la conformación de una proteína que tiene un acido glutámico formando una alfa hélice, su energía dependerá en parte de cuan probable es esa conformación para el glutámico. La probabilidad de la conformación alfa hélice para el glutámico se calcula evaluando cuántas veces este aminoácido se encuentra en esa conformación sobre el total de conformaciones observadas en todas las estructuras de proteínas resueltas experimentalmente. En este caso, la bioinformática estructural se dedica a la generación y análisis de bases de datos y a la posterior construcción de los potenciales estadísticos. Estos potenciales tuvieron su auge en los últimos años con las competencias llamadas CASP (Critical Assessment of Structure Prediction), donde los participantes deben predecir la estructura de una proteína partiendo únicamente de su secuencia. La estructura “real” de dicha proteína ha sido determinada previamente por técnicas experimentales y al final de la competencia, se califica las predicciones realizadas de acuerdo a su similitud con la estructura “real”.
Los potenciales estadísticos han tenido gran uso para el estudio de la interacción droga-proteína dado que no siempre se cuenta con la estructura de la proteína blanco. Al evaluar la factibilidad de una droga se genera inicialmente un modelo de la proteína utilizando, de ser posible como punto de partida, la estructura conocida de una proteína homóloga. Los programas más conocidos, que realizan estos “modelos por homología” son el Modeller desarrollado en el laboratorio de Andreas Sali en la universidad de California en San Diego y el Swiss-Model actualmente desarrollado por el grupo de bioinformática estructural dirigido por Torsten Schwede en la universidad de Basilea.
La bioinformática estructural como eje del desarrollo de nuevos fármacos
El paradigma del descubrimiento de nuevas drogas o fármacos consiste en encontrar una molécula pequeña capaz de unirse e inhibir/activar eficientemente un blanco macromolecular específico, generalmente una proteína. La característica principal que debe poseer una droga es la de unirse fuertemente a la proteína blanco. La unión droga/inhibidor-proteína es caracterizada químicamente mediante la constante de inhibición (Ki):
Donde [P], [I] y [PI] son las concentraciones de proteína, inhibidor y el complejo proteína-inhibidor, respectivamente. La Ki está directamente relacionada con la energía de unión entre la proteína y la droga (ΔG) de acuerdo a la siguiente ecuación:
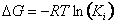
Las drogas que llegan a hacerse comerciales y poseen valor terapéutico, tienen por lo general valores de Ki < 1nM (nanoMolar). Otra manera de caracterizar la potencia de una droga es la medición del IC50, que es la concentración de un inhibidor requerida para reducir a la mitad la actividad de la proteína en estudio. Por lo general IC50 ≈ Ki.
Identificación del Blanco Molecular
Actualmente, la mayoría de los programas de descubrimiento de drogas comienzan con la identificación de un blanco molecular con potencial terapéutico. Uno de los problemas es identificar posibles zonas de unión de una droga en la molécula blanco (druggability). Mediante el análisis de la estructura proteica diversos algoritmos permiten determinar la existencia de bolsillos hidrofóbicos capaces de unir compuestos tipo-droga (drug-like). En los casos en que la estructura de la proteína blanco es desconocida, como se mencionó anteriormente, se puede generar un modelo de la misma. Muchas proteínas contienen sitios de unión por necesidad, como los sitios activos en las enzimas, pero otras veces es posible bloquear la actividad proteica mediante la interacción con sitios alostéricos. Drogas exitosas del tipo alostérico son por ejemplo los inhibidores de proteasas no-nucleosídicos para el tratamiento del HIV, como Efavirenz, Evirapine y Delavirdine.
Identificación de compuestos líderes (lead compounds)
Un compuesto líder es aquella molécula con un IC50 de aproximadamente 10µM contra la proteína blanco. En la actualidad, en la búsqueda de compuestos líderes, se utilizan robots que prueban el efecto de miles de drogas en un ensayo previamente estandarizado para determinar su eficacia. Este tipo de ensayos, denominados high-throughput screening assays (HTP), son extremadamente costosos y requieren poseer una biblioteca de posibles fármacos, y desarrollar además un ensayo eficiente para determinar su actividad. Aquí la utilización de técnicas in-silico permite reducir los costos y acelerar el proceso de búsqueda de manera significativa. Las técnicas in-silico para detectar compuestos lίderes responde a tres tipos de estrategias. La primera, denominada búsqueda de similitud (“similarity search”), se basa en la idea de que proteínas con estructura similar suelen unir compuestos parecidos. Utilizando técnicas de alineamiento estructural se buscan proteínas con poca identidad de secuencia (menor al 30%) pero estructuralmente similares a la proteína blanco, y para las cuales se conozcan inhibidores efectivos. Comparando ambas estructuras y sus inhibidores, es posible seleccionar compuestos líderes a partir de estos candidatos.
La segunda estrategia para la identificación de compuestos líderes se basa en la realización de una búsqueda virtual de moléculas capaces de unirse a la proteína blanco, a partir de una biblioteca de compuestos drug-like (una suerte de biblioteca o enorme archivo de compuestos químicos), en lo que se conoce como Virtual Screening. Las bibliotecas de compuestos farmacológicos pueden ser reales, es decir representar una colección de compuestos provistos por una compañía farmacéutica o química, o virtuales, generados a partir de fragmentos moleculares. La biblioteca ZINC, que es gratuita y de libre acceso, tiene más de 8 millones de compuestos y se pueden obtener las estructuras 3D para realizar experimentos virtuales.
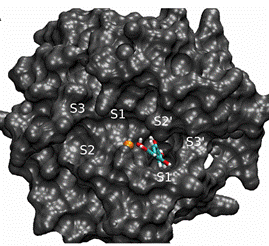
El método más utilizado para el virtual screening es el denominado “Docking”. El mismo consiste en encontrar la “mejor” manera de acomodar la droga en estudio dentro de la estructura de la proteína blanco. Para ello se utilizan algoritmos que realizan una rápida búsqueda de conformaciones las cuales son evaluadas mediante una función que les asigna un puntaje (Scoring function) relacionado directamente con su afinidad (Figura 1). Los métodos para calcular la afinidad en un programa de docking se basan en alguna estimación de la energía libre de unión mediante la realización de un cálculo aproximado para las distintas contribuciones, que comprenden: la energía de interacción electrostática, la energía de Van der Waals (dada por la complementariedad de forma y superficie) y el cambio en la energía libre de solvatación asociado principalmente con la solubilidad del fármaco. Aquellas drogas para las cuales se predice una alta afinidad son seleccionadas para estudios experimentales con la esperanza de que se conviertan en compuestos líderes. Los programas más conocidos utilizados para realizar este tipo de estudios son el Auto Dock, el Dock y el FlexX, entre otros.
Finalmente la tercera estrategia, denominada Diseño De-novo, consiste en armar el fármaco combinando fragmentos moleculares seleccionados para interactuar con las diferentes zonas del sitio de unión. De más esta decir que es posible combinar las tres estrategias, por ejemplo realizando un screening virtual sobre un conjunto de compuestos seleccionados por su capacidad de unirse a una proteína relacionada con el blanco.
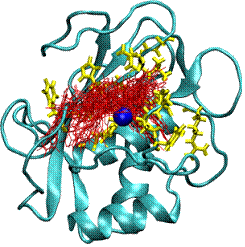
Figura 1. (A) Búsqueda conformacional la posible interacción entre una droga (Rojo) y la proteína blanco.
En este caso la proteína blanco es una metalo enzima que contiene Zn (Azul). Los residuos del sitio activo de la proteína se muestran en amarillo. (B) Estructura del complejo droga-enzima con mayor puntaje. Nótese la inclusión del fármaco en un bolsillo y su interacción con el Zn (naranja).
Optimización de compuestos líder
Una vez identificado un compuesto líder, se procede a la fase de optimización que consiste en aumentar su afinidad por la proteína blanco hasta valores de Ki o IC50 <nM (subnanomolar). Este proceso, conocido como Structure-Based Drug Design (SBDD), ha sido exitoso en numerosos casos y ha contribuido con la introducción de más de 50 drogas a ensayos clínicos. Las etapas de análisis y propuestas de modificaciones a realizar sobre el compuesto inicial se llevan a cabo in-silico utilizando las diversas técnicas de la bioinformática estructural descriptas previamente.
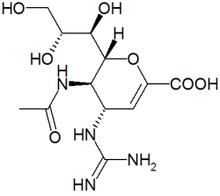
Un ejemplo significativo de SBDD, es el desarrollo del antiviral Relenza comercializado por Glaxo para combatir la gripe estacional. El blanco molecular de Relenza, también conocido como Zanamivir, es la neuraminidasa viral, de los virus influenza tipo A y B. El producto fue aprobado por la FDA en 1999 y se encuentra registrado en 70 países. Recientemente en el 2006 Alemania anunció la compra de 1.7 millones de dosis de Relenza como posible estrategia para combatir la gripe aviar. El desarrollo de Zanamivir fue llevado a cabo por el grupo liderado por el Dr Mark von Itzstein del Victorian College of Pharmacy, Monash University. A partir de la estructura proteica de la neuraminidasa viral, determinada por cristalografia de rayos-X, y el conocimiento de un inhibidor moderado, el 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA) se realizó una búsqueda in-silico de modificaciones que aumentaran su potencia. El análisis del potencial electrostático del sitio activo de la neuraminidasa mostró una zona negativa cercana al grupo oxidrilo-C4 del DANA. En base a este resultado se decidió modificar el sustituyente en esta posición con un grupo positivo. La propuesta resultante fue la incorporación de un grupo tipo guanidinio con carga positiva capaz además de formar un puente salino con el Glutámico 119, uno de los aminoácidos más conservados del sitio activo. El resultante Zamanivir resultó más de 100 veces más potente que el precursor.
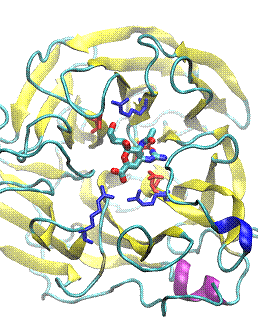
Figura 2: (A) Zanamivir (B) Estructura del complejo Neuraminidasa-Zanamivir. En azul se resaltan dos argininas que interactúan con el grupo carboxilato y en rojo se resalta el Glutámico 119 que interactúa con el grupo guanidinio del Zanamivir.
Conclusión
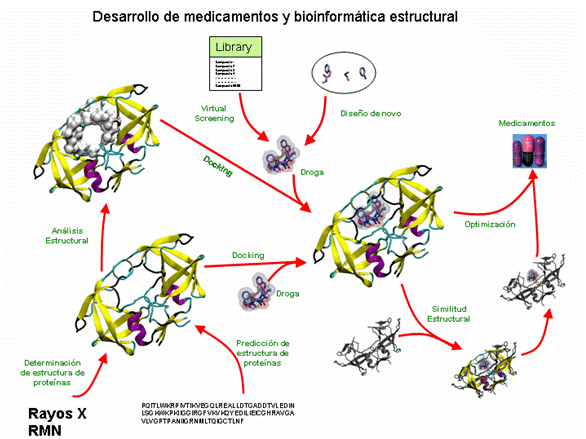
En resumen, la bioinformática estructural participa actualmente de manera significativa en las diversas etapas del desarrollo de nuevos fármacos. Dado que cada una de estas etapas requiere de la realización de costosos experimentos, aquellos grupos o compañías que han incorporado herramientas informáticas a sus programas de desarrollo, adquieren la ventaja de llegar al objetivo más rápido y a un menor costo que sus competidores.
Figura 3: Etapas del desarrollo de un medicamento y la contribución de la bioinformática estructural en cada paso.
¿Por qué enseñar y aprender bioinformática estructural?
Hoy en día la mayoría de la comunidad de biólogos moleculares y bioquímicos considera que la bioinformática consiste sólo o principalmente en el estudio y análisis de bases de datos de secuencias genómicas y de proteínas. Esta visión se transmite a los alumnos que se acercan a tomar cursos de bioinformática buscando aprender solamente algoritmos de comparación de secuencias (BLAST), o a realizar búsquedas en bases de datos (Usar el Entrez del sitio de NCBI (http://www.ncbi.nlm.nih.gov/Database/index.html ). Sin embargo estas herramientas son sólo la punta del iceberg de la bioinformática, que cuando se combina con la creciente información estructural y el conocimiento de las bases físico-químicas que gobiernan el comportamiento de los sistemas biológicos a nivel atómico y molecular, se convierte en una herramienta de proyecciones inimaginables. En este contexto, es indispensable formar a los biólogos o bioquímicos o químicos del futuro con interés en la biologίa molecular o estructural, con una sólida base en ciencias (Física, Química, Matemática y Computación) y un conocimiento general de las herramientas y fundamentos de la bionformática estructural.
Por otro lado desde el punto de vista del alumno, es importante notar que la adquisición de los conceptos y habilidades de bioinformática estructural le permitirán, no sólo ampliar sus horizontes de investigación, sino convertirse en una posible salida laboral en el ámbito profesional, como lo demuestra la existencia de numerosas compañías abocadas al desarrollo de fármacos in-silico.
Referencias
Philip E. Bourne and Helge Wissig, Structural Bioinformatics. 2003, Wiley-Liss de. New Jersey.
W. L. Jorgensen, The Many Roles of Computation in Drug Discovery. Science, 2004; (303) 1813-1818.
D.B Kitchen et. al., Docking and Scoring in Virtual Screening for Drg Discovery: Methods and Applications. Nature Reviews Drug Discovery, 2004; (3) 935-949.
M, von Itzstein et. al. , Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993; (363) 418-423.
H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne The Protein Data Bank. Nucleic Acids Research, 2000. (28). 235-242.
ISSN 1666-7948
www.quimicaviva.qb.fcen.uba.arRevista QuímicaViva
Número 1, año 8, Abril 2009
quimicaviva@qb.fcen.uba.ar