Prospectiva del uso de esteroides de plantas como antivirales
Viviana. Castillaa*, Javier Ramírezb y Celia E. Cotoa
a Laboratorio de Virología, Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina
b Departamento de Química Orgánica y UMYMFOR, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina.
E mail: viviana@qb.fcen.uba.ar
Recibido el 30/03/09. Aceptado el 10/04/09.
Resumen
Las plantas sintetizan una gran variedad de esteroides, algunos de ellos poseen función hormonal y otros participan en los mecanismos de defensa frente a la infección con microorganismos patógenos. En la presente actualización se analiza el potencial de este grupo de compuestos como agentes antivirales frente a virus que infectan al hombre o a los animales. En particular se comenta el espectro de acción de diversos esteroides naturales y sus derivados sintéticos, la influencia de cambios estructurales en la actividad antiviral y los mecanismos de acción propuestos hasta el momento.
Palabras clave: esteroide, antiviral, virus, brassinoesteroide, seco-pregnano, ácido ursólico
Prospective use of plant steroids as antiviral compounds
Abstract
Plants possess tha ability of synthesize a large variety of steroids, some with hormonal functions and others involved in defense mechanisms against microbial pathogens. In the present review we discuss the potential of plant steroids as a promissory group of compounds with antiviral properties against human and animal viruses. The analysis will fucus on the antiviral spectrum of natural and synthetic steroids, the influence of structural changes on antiviral activity and the mechanisms of action that have been proposed up-to-date.
Key words: steroid, antiviral compound, virus, brassinosteroid, seco-pregnane, ursolic acid
Introducción.
Desde los tiempos primigenios, en los que su naturaleza se desconocía, hasta su aislamiento e identificación a principios del siglo XX, los virus constituyen un flagelo para el hombre, animales y vegetales; incluidos mamíferos, insectos, peces, reptiles, plantas y organismos unicelulares como bacterias, parásitos y hongos. Son parásitos obligados pero también tienen una etapa extracelular que les permite saltar de huésped en huésped, por ello, son capaces de producir enfermedades agudas, no siempre mortales, pero insidiosas. Generalmente perviven en forma de infecciones persistentes cohabitando con el huésped que parasitan en forma silenciosa o con períodos de recurrencia activa.
El diseño y aplicación masiva de algunas vacunas ha permitido poner un freno a algunos virus causales de epidemias como el virus polio o el de la viruela. Pero los virus, omnipresentes en todos los seres que habitan la tierra, de pronto se manifiestan como si fueran nuevas entidades originando graves problemas sanitarios como ocurrió con la aparición del virus HIV y con un ejemplo actual en la Argentina: la epidemia de Dengue. La emergencia y la reemergencia de las enfermedades infecciosas es un mal de este siglo que hizo su eclosión a fines del siglo pasado.
Resulta fácil inferir que no es fácil combatirlos. No sólo por su gran abundancia y diversidad sino también porque existen numerosos serotipos de la misma especie y porque mutan constantemente. Los virus además de producir infecciones agudas pueden también ser causa de tumores y muchos de ellos forman parte de nuestro patrimonio genético como los retrovirus y otros segmentos genéticos derivados.
¿Cómo luchar contra los virus? Mencionamos antes la existencia de vacunas pero no hay vacunas para todos los virus y entre las que existen no todas son verdaderamente eficaces. Otra forma de lucha es desarrollar drogas capaces de inhibir la replicación viral conocidas como antivirales. Lamentablemente, a diferencia con las bacterias es difícil obtener un antiviral de amplio espectro ya que hay muchos grupos de virus (dependiendo del tipo de genoma que poseen) cuyo ciclo de replicación es diferente. Esta situación es muy limitante a lo que debemos agregar que es vital que la droga que se obtenga no debe afectar la bioquímica celular, en otras palabras; debe carecer de citotoxicidad para el huésped.
De los miles de compuestos ensayados tanto naturales como sintéticos sólo menos de una docena se encuentran en uso clínico por eso la búsqueda de nuevas estructuras químicas con potencial antiviral no cesa. Uno de los caminos a investigar son las plantas. En un trabajo reciente aparecido en QuímicaViva Número 2, año 6, agosto 2007 sobre “Plantas sanadoras: Pasado, presente y futuro”, su autora Andrea A. Barquero describe en detalle la historia de los productos sanadores obtenidos de las plantas y presenta un cuadro detallado de todos los medicamentos en uso derivados de ellas.
A finales de la última década del siglo XX se comenzó a investigar una nueva familia de compuestos de estructura química diferente a los antivirales conocidos: los esteroides. Estos compuestos poseen un esqueleto de ciclopentano [a]fenantreno o un esqueleto derivado a partir del mismo tanto como por contracción o expansión de los anillos o por ruptura de algún enlace. Normalmente están presentes también grupos metilo en los carbonos 10 y 13, y una cadena alquílica en el carbono 17.
Los esteroles son esteroides que poseen un grupo hidroxilo en el carbono 3.
Cientos de esteroides distintos se encuentran en plantas, animales y hongos. Asimismo, numerosos derivados de esteroles fueron obtenidos por síntesis química y ensayados in vivo e in vitro frente a diversos virus patógenos para el hombre.
Algunos esteroides de origen animal se aplican en la clínica de las enfermedades virales con éxito dispar. En la actualización presente también nos referimos al potencial de esta familia de moléculas pero nos ocuparemos sólo de los compuestos naturales derivados de plantas y de los obtenidos por síntesis, y cuya estructura química se puede considerar de naturaleza esteroidal.
Esta actualización consta de dos partes: a) estudios realizados en nuestro laboratorio con brasinoesteroides naturales y sintéticos y b) la acción de compuestos de estructura relacionada estudiados en otros laboratorios de investigación
Esteroides derivados de las plantas
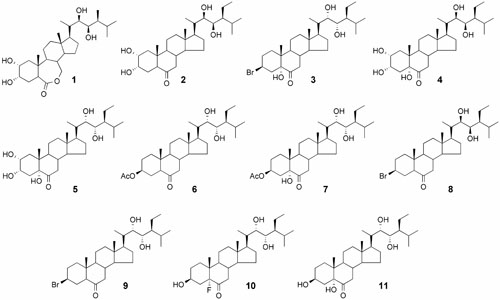
Las plantas pueden sintetizar una gran variedad de esteroides, a este tipo de moléculas al igual que para los esteroides de origen animal, se les ha asignado una función hormonal. El primer esteroide con actividad hormonal aislado de una fuente natural fue el brassinólido (1) (Fig. 1), en 1979 a partir del nabo silvestre Brassica napus.
Fig. (1): Brassinoesteroides naturales y sintéticos
Posteriormente se aislaron numerosos compuestos esteroidales de plantas, estructuralmente relacionados, que promueven el crecimiento vegetal. Se conocen alrededor de 60 compuestos (Bajguz, A., 2003) que colectivamente se denominan brasinoesteroides (BRs). Estas moléculas contienen el esqueleto esteroidal típico de 5a-colestano con anillos fusionados A, B, C y D y una cadena lateral alquílica que nace en el C-17 que no se encuentra en las hormonas esteroidales de mamíferos.
Los BRs son esteroides ampliamente distribuidos en las plantas y funcionan como moléculas señal involucradas en procesos como la elongación del tallo, la diferenciación vascular, fertilidad masculina, tiempo de senescencia y florecimiento, desarrollo de las hojas y resistencia al estrés biótico y abiótico. De forma similar a los esteroides animales con función hormonal, los BRs regulan la expresión de numerosos genes, impactan en la actividad de vías metabólicas complejas y ayudan al control del desarrollo y la morfogénesis (Ryu et al., 2007).
La presencia de los BRs es percibida por la membrana plasmática por unión directa con los dominios extracelulares de la proteína transmembrana BRII, que tiene actividad de serina-treonina proteína-quinasa. La unión del BR inicia una cascada de señales de modo que otras quinasas y fosfatasas determinan la fosforilación y estabilidad de los factores de transcripción nucleares. Esos factores son los que median los efectos mayores de los BRs en los varios procesos fisiológicos de las plantas (Karlova y de Vries, 2006).
Las plantas también han desarrollado mecanismos de defensa constitutivos e inducidos sintetizando un gran conjunto de metabolitos secundarios contra patógenos microbianos
(Dixon, 2001).
En consecuencia, es probable que los compuestos antivirales de las plantas sean parte de su defensa innata y por lo tanto constituyan un grupo promisorio de compuestos para la búsqueda de nuevos agentes antivirales.
Hormonas: brassinoesteroides
A fines de la década del 90 nuestro laboratorio de Virología comenzó el estudio de BRs sintéticos y naturales provistos por el grupo de Química Orgánica dirigido por la Dra. Lydia Galagovsky, con la participación del Dr. Javier Ramírez, con el propósito de investigar si estas moléculas poseían actividad antiviral frente a virus animales cultivados in-vitro. Así es como se encontró que varios BRs sintéticos y naturales poseían una acción inhibitoria sobre la replicación de diferentes virus (Wachsman et al., 2000; Wachsman et al., 2002; Talarico et al., 2002; Wachsman et al., 2004a; Wachsman et al., 2004b). Primero se demostró que dos BRs naturales (1) y la 28-homocastasterona (2) (Fig. 1) mostraban actividad antiviral contra poliovirus, herpes simplex virus (HSV-1), virus de la estomatitis vesicular (VSV) y los arenavirus Junín (JUNV) y Tacaribe (TCRV) (Wachsman et al., 2004b). Los valores de inhibición reportados fueron superiores a los publicados para otros esteroides ensayados previamente (Koehn et al., 1991; Comin et al., 1999; Arthan et al., 2002). Después de esos estudios preliminares se evaluó la actividad citotóxica y antiviral de una serie de derivados sintéticos de BRs (3-11) (Fig. 1) obtenidos mediante síntesis química a partir del estigmasterol un esterol natural de plantas. La mayoría de los compuestos ensayados mostraron una mejor actividad que la 28- homocastasterona (2) que posee el esqueleto esteroidal básico de todos los compuestos sintéticos. En el cuadro 1 se muestra la actividad antiviral de algunos derivados expresada en términos de índice de selectividad (IS), que resulta del cociente entre el valor CC50 (concentración de compuesto requerida para reducir en un 50% la viabilidad celular) y el valor CE50 (concentración de compuesto requerida para reducir en un 50% la replicación viral).
Los compuestos 3, 10 y 11 resultaron activos contra la mayoría de los virus ensayados: HSV-1, HSV-2, virus del sarampión (MV) y JUNV. El compuesto más activo fue el 3, sin embargo este compuesto resultó menos activo que el antiviral aciclovir (ACV) contra HSV-1 y HSV-2 (Talarico et al., 2002; Wachsman et al., 2004b). Por el contrario, este compuesto mostró una actividad de 3,5 a 18 veces mayor que la ribavirina contra los virus MV y JUNV respectivamente (Wachsman et al., 2002; Wachsman et al., 2004b). Los derivados de los BRs fueron muy efectivos para inhibir la infección in vitro con JUNV con valores de IS mayores de 100 para los compuestos 3 al
8. Resultados similares se obtuvieron con otros arenavirus como TCRV y el virus Pichinde lo que indica que los miembros de esta familia de virus son muy sensibles a este tipo de compuestos (Wachsman et al., 2000).
Con la idea de encontrar una relación estructura química-actividad antiviral se probaron más 30 compuestos análogos. Los BRs naturales, por ejemplo, la 28-homocastasterona (2) es una fitohormona muy activa que posee un diol con una configuración 22R, 23R en la cadena lateral.
Cuadro 1: Actividad antiviral de brasinoesteroides (BRs)
Compuesto
CC50 (µM)
IS
HSV-1
HSV-2
MV
JUNV
2
63
2
ND
2
18
3
277
100
71
44
693
4
263
1
ND
2
200
5
502
16
ND
6
228
6
226
10
ND
8
232
7
462
17
ND
20
231
8
114
4
ND
2
310
9
152
6
ND
4
72
10
160
109
27
54
27
11
1044
80
40
40
40
CC50: concentración de compuesto requerida para reducir en un 50% la viabilidad de las células Vero respecto al control sin tratar con compuesto. IS: índice de selectividad o cociente entre los valores de CC50 y de CE50. CE50: concentración de compuesto requerida para reducir en un 50% el rendimiento viral respecto a un cultivo celular infectado no tratado con el compuesto (Wachsman et al., 2004b).
Según los estudios realizados encontramos que los análogos que tienen la configuración opuesta, no natural, en esa posición muestran generalmente una actividad antiviral exacerbada, comparar 4 con 5 o 9 con 8. Por otra parte, la introducción de un grupo electronegativo en C5 (flúor en 10 e hidroxilo en 11) origina compuestos más activos contra el HSV-1 (Cuadro 1).
Como ninguno de los compuestos ensayados mostró un efecto de inactivación directa sobre las partículas virales se presupone que su acción se ejerce en un paso del ciclo de replicación viral. Con el objetivo de determinar el paso de replicación afectado se trabajó con el compuesto 3 y el virus HSV-1, encontrándose que en presencia del BR la síntesis tardía de las proteínas virales estaba afectada severamente (Wachsman et al. 2004a.). Dado que la síntesis de proteínas tardías depende de que haya ocurrido la síntesis de ADN viral, se realizaron estudios usando el BR 3 en combinación con ACV o foscarnet (FOS), éstos últimos antivirales conocidos por su efecto inhibitorio de la síntesis de ADN viral. Se encontró que el BR 3 actúa en forma sinérgica con concentraciones bajas de ACV y moderadas de FOS (Talarico et al., 2006). Estos resultados sugieren que el mecanismo de acción antiviral del compuesto 3 difiere de los antivirales dirigidos a impedir la síntesis del ADN viral.
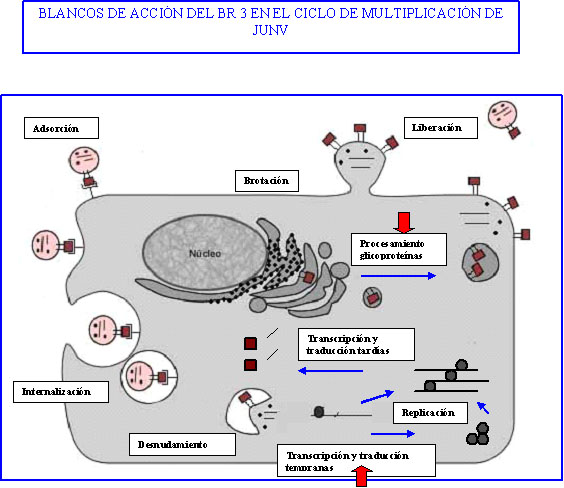
El modo de acción del compuesto 3 contra la replicación de JUNV en células Vero se investigó con el objeto de determinar el paso afectado (Castilla et al., 2005). para lograr una mayor comprensión del proceso de replicación se muestra aquí un diagrama esquematizado de la replicación del virus Junín y los posible blancos de ataque de un antiviral.
Las flechas rojas indican los posibles blancos de acción del BR 3 en el ciclo de multiplicación de JUNV
La realización de experimentos de agregado del compuesto 3 a distintos tiempos del ciclo de replicación viral reveló que este compuesto fue más efectivo si se agregaba durante el período temprano de la infección. Además el BR 3 previno la formación de partículas virales maduras. En concordancia con los resultados obtenidos previamente con HSV-1 no estaba afectada ni la adsorción ni la penetración del virus a las células. En el caso de JUNV, el paso subsiguiente a la entrada del virus a la célula es la síntesis del ARN viral, por esa razón se realizaron experimentos encaminados a esclarecer el efecto del compuesto en la síntesis de ARN viral. El genoma del virus Junín está compuesto de dos fragmentos denominados S y L cuya estrategia de replicación ocurre en ambos sentidos. Esto quiere decir que parte del genoma se expresa como si fuera un ARN positivo y la otra parte como un ARN negativo. El fragmento S, que codifica por la proteína de la nucleocápside (N) y el precursor de las glicoproteínas virales, es transcripto a dos formas antigenómicas: un ARN mensajero de la proteína N de 1.8 kb (transcripción primaria) y un ARN de longitud completa antigenómico de S de 3,4 kb (replicación del genoma); una estrategia similar se ha descripto para el fragmento L. El análisis mediante la técnica RT-PCR (reacción de la polimerasa por retrotranscripción) mostró que en los cultivos infectados y tratados con el compuesto 3 sólo se encontraba el ARN mensajero de N indicando que la presencia del BR prevenía la síntesis del ARN viral antigenómico.
Se ha propuesto que el paso de la transcripción a la replicación en el proceso de infección de los arenavirus depende del nivel de acumulación intracelular de la proteína N ya que se ha demostrado que en condiciones de niveles bajos de N sólo se producen transcriptos de ARN mensajero de N. La idea es que al traducirse el ARN mensajero la acumulación de proteína N actuaría como un factor antiterminador transcripcional, de esta manera tiene lugar la síntesis completa del ARN S antigenómico (Meyer, 2002).
En consecuencia, se puede postular que el compuesto 3 estaría bloqueando ya sea la síntesis de proteína N o su función. Sin embargo, lo que ocurre verdaderamente es difícil de saber ya que el cambio de la transcripción a la replicación en los arenavirus puede resultar más complejo e involucrar a otros factores celulares y virales. Además del efecto sobre la síntesis del ARN se pudo observar que el BR 3 produce una inhibición importante de los rendimientos de virus y de su poder de fusión de las células cuando se lo agregó al final del ciclo de replicación. Dados estos resultados no se puede evitar pensar que el compuesto 3 tenga un efecto adverso en el procesamiento pos-traducción de las glicoproteínas virales o alternativamente que afecte la inserción correcta de las glicoproteínas virales en la membrana celular.
Se investigó también el efecto del BR 3 en los diferentes pasos del ciclo replicativo del virus VSV (Romanutti et al., 2007). En este modelo de replicación de un virus ARN negativo de genoma de una sola hebra, la transcripción primaria no se inhibió pero, por el contrario, se encontró una alta inhibición de la síntesis de proteínas virales y de la formación de las partículas maduras. A pesar de que el efecto de 3 no se estudió todavía sobre la síntesis del ARN completo, los resultados obtenidos están en concordancia con los relatados en relación a JUNV.
Si consideramos que el compuesto 3 afecta a virus de naturaleza tan dispar como los herpes (ADN de cadena doble), sarampión y VSV (ARN cadena negativa de una hebra) y arenavirus (ARN positivo-negativo de una hebra, segmentado) podemos concluir que estamos frente a un antiviral de amplio espectro. El hecho de que algunos virus sean más susceptibles que otros podría estar indicando que el compuesto afecta algún paso específico viral y en menor grado a una función celular requerida para la replicación viral.
Existen evidencias de que los BRs naturales tienen muy bajo nivel de citotoxicidad, son constituyentes de la mayoría de las plantas y usualmente son consumidos por los mamíferos. La falta de toxicidad de los BRs naturales se ha confirmado en estudios en ratones y ratas tratadas en forma oral o en forma dérmica (Khripach et al., 2000).
Datos provenientes de nuestro laboratorio indican que en estudios realizados in-vivo en un modelo experimental murino no se encontró efecto tóxico administrando el BR 3 (40μM) tres veces por día durante tres días consecutivos en forma tópica en los ojos de ratones adultos (Michelini et al., 2004).
Una de las pruebas obligadas de los antivirales es demostrar su efectividad en animales antes de realizar ensayos en humanos. Para el caso de los BRs se realizó una evaluación de su capacidad anti-herpética in vivo contra la queratitis estromal herpética (HSK) utilizando los compuestos 3, 10 y 11 suministrando éstos en los ojos de los animales durante los tres primeros días después de la infección. Se observó que la infección se retrasó y redujo su incidencia a pesar que los títulos de virus en los lavados oculares no disminuyeron en los animales tratados respecto de los no tratados. Lo que sugirió que los compuestos no funcionaban como verdaderos antivirales sino que disminuían la inflamación estromal de naturaleza inmune causada por la infección (Michelini et al., 2004). Esta hipótesis de trabajo fue reforzada posteriormente por estudios in-vitro en los que se demostró que el BR 3 modulaba la respuesta de las células epiteliales e inmunes a la infección con HSV-1 ya sea actuando como inductor o inhibidor de la producción de citoquinas dependiendo del tipo celular involucrado (Michelini et al., 2008a). En consecuencia, el efecto protector observado en los ratones puede interpretarse como un balance entre los efectos inmunoestimulantes e inmunosupresores de los compuestos. Resultados similares se obtuvieron con otros estigmastanos sintéticos. (Michelini et al., 2008b). Además, varios compuestos sintéticos derivados del estigmastano inhibieron la producción del factor de necrosis tumoral TNF-α en células L929. Según análisis preliminares de relación estructura-actividad, el efecto modulador sobre la producción de TNF-α de estos compuestos sintéticos puede estar relacionado con la presencia de la cadena lateral de estigmastano dihidroxilada con configuración 22S, 23S y con un sistema 3β,5α-dihidroxy-6-ceto en los anillos A y B del esteroide. (Ramírez et al., 2007).
Es interesante recalcar que tanto la actividad antiviral como las propiedades inmunomoduladoras de los compuestos estudiados parecen requerir la presencia de la cadena lateral alquílica (que no se encuentra en los esteroides de mamíferos) y que además esta cadena esté dihidroxilada en los carbonos 22 y 23, con una estereoquímica opuesta a la que se encuentra en los brassinosteroides naturales. Notablemente, los compuestos que resultaron más activos como antivirales tienen una muy baja actividad hormonal en plantas.
Metabolitos secundarios: secopregnanos y otros esteroides
Esteroides seco-pregnanos
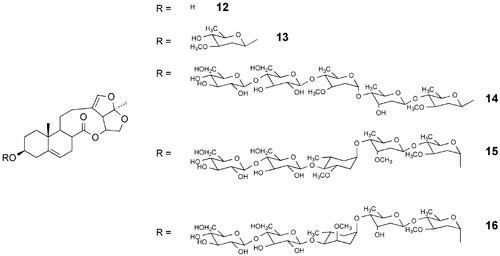
Estudios recientes aportaron evidencias que los esteroides seco-pregnanos y sus glicósidos, aislados de plantas herbáceas como Strobilanthes cusia and Cynanchum paniculatum, son efectivos e inhibidores selectivos de la replicación de varios miembros de la familia de los alfavirus. Estos son virus de ARN de sentido positivo e incluyen virus que infectan plantas y animales (Li et al., 2007). Con el objetivo de rastrear la presencia de compuestos antivirales en plantas se usó como virus modelo o blanco el virus del mosaico del tabaco (TMV). Mediante esta estrategia se logró el aislamiento de cinco esteroides seco-pregnanos con actividad anti-TMV a saber: la glaucogenina C (12) y el cynatrósido A (13)(aislados de S. cusia) y los paniculatumósidos C (14), D (15) y E (16) aislados de C. paniculatum (Fig. 2). Posteriormente se evaluó su acción sobre la replicación del togavirus Sindbis cuya organización genómica y estrategia de replicación es semejante a la del TMV. Los cinco compuestos inhibieron la cipatogenicidad inducida por el virus Sindbis en células BHK21 sin efectos tóxicos sobre la célula ni sobre su proliferación. El compuesto 14 resultó ser también un efectivo inhibidor de otros alfavirus. Por el contrario, este compuesto no afectó la multiplicación de otros virus ARN o ADN (Cuadro 2) señalando que su acción es muy específica.
La actividad antiviral de los cinco compuestos (12 al 16) (Fig. 2) fue similar indicando
que la aglicona per se es responsable del efecto inibitorio y que el las cadenas de oligosacáridos contribuyen poco a la acción antiviral.
Fig. (2): Seco-pregnanos naturales aislados a partir de S. cusia (12 and 13) y C. paniculatum (14, 15 and 16)
Cuadro 2: Espectro antiviral del esteroide seco-pregnano paniculatumósido C (14)
Virus
Familia
Células
CE50 (nM)
IS
SINV
Togaviridae
BHK-21
1,5
11.000
GETV
Togaviridae
BHK-21
1
16.500
EEV
Togaviidae
BHK-21
2
8.250
JEV
Flaviviridae
C6/36
>24.000
I
HCV
Flaviviridae
B
>24.000
I
HIV
Retroviridae
H9
>24.000
I
MV
Paramyxoviridae
Vero
>24.000
I
virus influenza A
Orthomyxoviridae
MDCK
>24.000
I
reovirus
Reoviridae
CIK
>24.000
I
adenovirus humano 4
Adenoviridae
983A
>24.000
I
CC50: concentración de compuesto requerida para reducir en un 50% la viabilidad celular respecto al control sin tratar con compuesto. CE50: concentración de compuesto requerida para producir un 50% de inhibición del efecto citopático inducido por el virus. IS: razón CC50/CE50. I: inactivo (Li et al., 2007).
El estudio del modo de acción de estos compuestos frente a SINV permitió establecer que los mismos afectan la síntesis de ARN viral (Li et al., 2007). Al comienzo de la infección con SINV se sintetizan dos ARN mensajeros, el ARN genómico (49S) a partir del cual se traducen las proteínas virales no estructurales y el ARN subgenómico (26S) que codifica para las proteínas estructurales del virus (la proteína de cápside y dos glicoproteínas de envoltura). La síntesis de ARN subgenómico se encuentra fuertemente inhibida en presencia de esta clase de esteroides mientras que la síntesis de ARN genómico no parece estar afectada (Li et al., 2007). Se ha propuesto que estos compuestos impiden la unión del complejo de transcripción a la cadena de ARN antigenómico utilizada como molde aunque no puede descartarse un posible efecto de los esteroides sobre factores celulares asociados a la maquinaria de transcripción.
La administración in-vivo del esteroide seco-pregnano 14 a ratones BALB/c recién nacidos los protege contra la infección letal con SINV (Li et al., 2007). Si los animales que sobrevivieron a la infección se vuelven a desafiar con una dosis letal de virus, no sobrevive ningún animal por lo cual se interpreta que el compuesto protege a los ratones por una inhibición directa de la replicación viral y no porque contribuye a montar una respuesta inmune protectora.
Ácido ursólico y compuestos estructuralmente relacionados
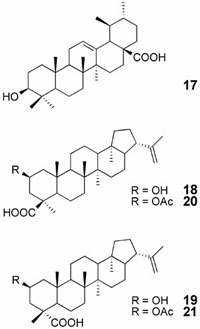
El ácido ursólico (ácido 3b-hidroxiurs-12-en-28-oico, 17), es un triterpenoide (Fig. 3) derivado de hojas de bayas, flores y frutas de varias plantas medicinales que despliega actividades biológicas varias: actividad antibacteriana, hepatoprotectora, inmunomoduladora, antiproliferativa y antiviral de amplio espectro.
Fig. (3): Ácido ursólico (17) y compuestos relacionados
Extractos y componentes purificados de Ocimum basilicu, una hierba medicinal utilizada en la medicina China, se utilizaron para detectar probables componentes antivirales contra virus ADN y ARN. Entre los virus ADN se probaron: HSV-1, adenovirus (ADV) y el virus de la hepatitis B y entre los virus ARN: coxsachievirus B1 (CV-B1) y enterovirus 71 (EV 71) (Chiang et al., 2005). Entre los compuestos activos, el ácido ursólico (17) mostró actividad antiviral contra HSV-1, ADV-8 y EV71 (Cuadro 3).
Cuadro 3: Actividad antiviral in vitro del ácido ursólico (17)
Virus
CE50 (mM)
IS
HSV-1
14,5
15,2
ADV-8
9,2
23,8
CVB1
0,9
251,3
EV71
1,1
201,0
HIV-1
4,4
3,3
CC50: concentración de compuesto que reduce en un 50% el crecimiento celular (células BCC-1/KMC como sustrato para la multiplicación de los virus HSV-1, ADV-8, CVB1 y EV71 o células H9 para la infección con HIV-1). CE50: concentración de compuesto requerida para reducir en un 50% el efecto citopático inducido por el virus en el caso de las infecciones con los virus HSV-1, ADV-8, CVB1 y EV71. En el caso de la infección con HIV la CE50 corresponde a la concentración de compuesto requerida para reducir en un 50% la cantidad de antígeno viral p24 presente en el sobrenadante de los cultivos en comparación con los cultivos control no tratados con el compuesto (Kashiwada et al., 2000; Chiang et al., 2005).
También se evidenció una ligera actividad del ácido ursólico contra HIV (Kashiwada et al., 2000). Investigaciones muy recientes demostraron que dos compuestos nuevos triterpenos del tipo hopano, conocidos como ácidos dioptérico A (18) y B (19) (Cuadro 4, Fig. 3) aislados del rizoma de Dryopteris crassirhizoma (Aspiadaceae) junto con el ácido ursólico muestran una actividad inhibitoria importante contra la proteasa del virus HIV. Además, la acetilación de los compuestos que da origen a los compuestos 20 y 21 incrementa en forma apreciable la actividad inhibitoria (Lee et al., 2008).
Fig. (3): Ácido ursólico (17) y compuestos relacionados
El ácido ursólico parece tener muy buenas perspectivas como antiviral de amplio espectro ya que también se ha reportado que su presencia suprime el crecimiento de células provenientes de un carcinoma cervical positivo para el virus del papiloma humano (HPV). La infección por HPV es la enfermedad de trasnsmisión sexual de mayor prevalencia en el mundo. Tiene lugar hasta en el 75% de las mujeres sexualmente activas, mientras que el 99,7% de los cánceres cervicales están asociados a infecciones previas con uno o más tipos oncogénicos del HPV. Este virus contiene como genoma un ADN doble circular que codifica por ocho genes de los cuales los denominados E6 y E7 son necesarios para la conversión maligna. La asociación de las proteínas de dichos genes con las proteínas supresoras de tumores p53 y pRB se ha propuesto como mecanismo por el cual dichos genes virales inducen tumores. Células HeLa portadoras de HPV-18 tratadas con ácido ursólico (17) desarrollan características típicas de apoptosis y presentan niveles menores de expresión génica de E6 y E7 determinado por la reacción de PCR reversa (Lee et al., 2008). Estos estudios sugieren que el ácido ursólico puede ser útil en el tratamiento de neoplasias cervicales asociadas al HPV.
Cuadro 4: Capacidad de compuestos aislados a partir de rizoma de Dryopteris crassirhizoma de inhibir la actividad de la proteasa de HIV-1
Compuesto
Nombre
CE50 (mM)
17
Ácido ursólico
8,9
18
Ácido dryoptérico A
26,5
19
Ácido dryoptérico B
44,5
20
Ácido 2-O-Acetildryoptérico A
1,7
21
Ácido 2-O-Acetildryoptérico B
10,8
CE50: concentración de compuesto que reduce en un 50% la actividad de la proteasa (PR) de HIV-1 respecto al control no tratado con el compuesto. La actividad inhibitoria se determinó usando el péptido sintético [His-Lys-Ala-Arg-Val-Leu-(pNO2-Phe)-Glu-Ala-Nle-Ser-NH2] como sustrato de una PR viral recombinante. El hidrolizado y el sustrato remanente se analizaron cuantitativamente por el método de HPLC (Lee et al., 2008).
La transcripción de los genes E6 y E7 del HPV está controlada por una región regulatoria río arriba denominada (URR). Factores transcripcionales participan en
la regulación positiva o negativa de los mismos, además se ha encontrado que la región URR contiene elementos de respuesta a esteroides como la progesterona y los glucocorticoides en base a lo cual se especula que en esa región existen elementos de respuesta al ácido ursólico que explicarían la regulación negativa de los genes de E6 y E7.
Nos encontramos así frente a propiedades más que interesantes de las moléculas con estructuras químicas de naturaleza esteroidal de origen vegetal.
Conclusiones
Los virus responden al tratamiento con antivirales con una selección rápida de partículas mutantes resistentes a la droga utilizada lo que obliga a los virólogos a la búsqueda de nuevos compuestos activos. Los esteroides obtenidos de fuentes naturales consisten en una gran variedad de compuestos que despliegan numerosas funciones, entre ellas, la modulación de la replicación viral. Los estudios centralizados en el uso de esteroides provenientes de mamíferos muestran un panorama complejo ya que si bien son activos in vitro, al aplicarlos a los pacientes no dejan de cumplir sus funciones hormonales lo que resulta perjudicial. (No se discutió en este trabajo pero existe abundante bibliografía).
Los esteroides de plantas no han sido estudiados en ensayos clínicos en humanos aunque hemos descripto su acción favorable en infecciones en modelos animales. Los ensayos in-vitro indican que a pesar de sus diferentes estructuras todos ellos ejercen una acción inhibitoria alterando esencialmente los procesos de transcripción/replicación viral.
En la figura 4 se han resumido las formas de acción que poseen los tres grupos de esteroides de origen vegetal discutidos en esta actualización. Sus acciones son específicas para diferentes grupos de virus y constituyen una reserva de compuestos que deberían ser ensayados en los casos en que fallan otros compuestos. Existe la esperanza de que, a diferencia con los esteroides animales, los esteroides de plantas no funcionen como hormonas y sí como antivirales. La carencia de efectos colaterales no deseados constituiría un requerimiento óptimo para utilizar en la clínica. Falta que algún laboratorio farmacéeutico se interese en esta clase de compuestos para realizar los ensayos correspondientes. Este tipo de compuestos ofrece además la posibibilidad de modificar por síntesis parte de la molécula en cuestión que posee efectos no deseados sobre las células del huésped.
Fig. (4): Mecanismos propuestos para la actividad antiviral de los esteroides de plantas.
Referencias
[1] Bajguz, A.; Tretyn, A. The chemical characteristic and distribution of brassinosteroids in plants. Phytochemistry, 2003, 62, 1027-46.
[2] Ryu, H.; Kim, K.; Cho, H.; Park, J.; Choe, S.; Hwang, I. Nucleocytoplasmic shuttling of BZR1 mediated by phosphorylation is essential in Arabidopsis brassinosteroid signaling. Plant Cell, 2007, 19, 2749-62.
[3] Karlova, R.; de Vries, S. C. Advances in understanding brassinosteroid signaling.
Sci. STKE, 2006, 354: pe36
[4] Dixon, R. A. Natural products and plant disease resistance. Nature, 2001, 411, 843-7.
[5 ] Wachsman, M. B.; López, E. M.; Ramírez, J. A.; Galagovsky, L. R.; Coto, C. E. Antiviral effect of brassinosteroids against herpes virus and arenaviruses. Antivir. Chem. Chemother., 2000, 11, 71-7.
[6] Wachsman, M. B.; Ramirez, J. A.; Galagovsky, L. R.; Coto, C. E. Antiviral activity of brassinosteroids derivatives against measles virus in cell cultures. Antivir. Chem. Chemother., 2002, 13, 61–6.
[7] Talarico, L. B.; Ramirez, J. A.; Galagovsky, L. R.; Wachsman, M. B.
Structure–activity relationship studies in a set of new brassinosteroid derivatives assayed against herpes simplex virus type 1 and 2 in cell cultures. Med. Chem. Res., 2002, 11, 434–44.
[8] Wachsman, M. B.; Castilla, V.; Talarico, L. B.; Ramirez, J. A.; Galagovsky, L. R.; Coto, C. E. Antiherpetic mode of action of (22S,23S)-3beta-bromo-5alpha,22,23-trihydroxystigmastan-6-one in vitro. Int. J. Antimicrob. Agents, 2004a, 23, 524-6.
[9] Wachsman, M. B.; Ramírez, J. A.; Talarico, L. B.; Galagovsky, L. R.; Coto, C. E.
Antiviral activity of natural and synthetic brassinosteroids. Curr. Med. Chem.- Anti-Infective Agents, 2004b, 3, 163-79.
[10] Kohen, F. F.; Gunasekera, M.; Cross, S. S. New antiviral sterol disulfate
ortho esters from the marine sponge Petrosia weinbergi. J. Org. Chem., 1991,
56, 1322–5.
[11] Comin, M. J.; Maier, M. S.; Roccatagliata, A. J.; Pujol, C. A.; Damonte, E. B.
Evaluation of the antiviral activity of natural sulfated polyhydroxysteroids and their synthetic derivatives and analogs. Steroids, 1999, 64, 335-40.
[12] Arthan, D.; Svasti, J.; Kittakoop, P.; Pittayakhachonwut, D.; Tanticharoen, M.; Thebtaranonth, Y. Antiviral isoflavonoid sulfate and steroidal glycosides from the fruits of Solanum torvum. Phytochemistry, 2002, 59; 459-63.
[13] Talarico, L. B.; Castilla, V.; Ramirez, J. A.; Galagovsky, L. R.; Wachsman M. B.
Synergistic in vitro interactions between (22S,23S)-3beta-bromo-5alpha,22,23-trihydroxystigmastan-6-one and acyclovir or foscarnet against herpes simplex virus type 1. Chemotherapy, 2006, 52, 38-42.
[14] Castilla, V.; Larzábal, M.; Sgalippa, N. A.; Wachsman, M. B.; Coto, C. E. Antiviral mode of action of a synthetic brassinosteroid against Junin virus replication. Antiviral Res., 2005, 68: 88-95.
[15] Meyer, B. J.; de la Torre, J. C.; Southern, P. J. In Arenaviruses: genomic RNAs transcription and replication; Oldstone, M. B. A., Ed.; Springer-Verlag: New York, 2002; Vol. 1, pp. 139–58.
[16] Romanutti, C.; Castilla, V.; Coto, C. E.; Wachsman, M. B. Antiviral effect of a synthetic brassinosteroid on the replication of vesicular stomatitis virus in Vero cells
Int. J. Antimicrob. Agents, 2007, 29, 311–6.
[17] Khripach, V.; Zhabinskii, V.; De Groot, A.Twenty years of brassinosteroids: steroidal plant hormones warrant better crops for the XXI century. Ann. Bot., 2000, 86, 441–7.
[18] Michelini, F. M.; Ramírez, J. A.; Berra, A.; Galagovsky, L. R.; Alché, L. E.
In vitro and in vivo antiherpetic activity of three new synthetic brassinosteroid analogues. Steroids, 2004, 69, 713-20.
[19] Michelini, F. M.; Berra, A.; Alché, L. E. The in vitro immunomodulatory activity of a synthetic brassinosteroid analogue would account for the improvement of herpetic stromal keratitis in mice. J. Steroid Biochem. Mol. Biol., 2008, 108, 164-70.
[20] Michelini, F. M.; Ramírez, J. A.; Berra, A.; Galagovsky, L. R.; Alché L. E. Anti-herpetic and anti-inflammatory activities of two new synthetic 22,23-dihydroxylated stigmastane derivatives. J. Steroid Biochem. Mol. Biol., 2008, 111, 111-6.
[21] Ramírez, J. A.; Bruttomesso, A. C.; Michelini, F. M.; Acebedo, S. L.; Alché, L.E.; Galagovsky, L. R. Syntheses of immunomodulating androstanes and stigmastanes: comparison of their TNF-alpha inhibitory activity. Bioorg. Med. Chem., 2007, 15, 7538-44.
[22] Li, Y.; Wang, L.; Li, S.; Chen, X.; Shen, Y.; Zhang, Z.; He, H.; Xu, W.; Shu, Y.; Liang, G.; Fang, R.; Hao, X. Seco-pregnane steroids target the subgenomic RNA of alphavirus-like RNA viruses. Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8083-8.
[23] Chiang, L.C.; Ng, L. T.; Cheng, P. W.; Chiang, W.; Lin, C. C. Antiviral activities of extracts and selected pure constituents of Ocimum basilicum. Clin. Exp. Pharmacol. Physiol., 2005, 32, 811-6.
[24] Kashiwada, Y.; Nagao, T.; Hashimoto, A.; Ikeshiro, Y.; Okabe, H.; Cosentino, L. M.; Lee, K. H. Anti-AIDS agents 38. Anti-HIV activity of 3-O-acyl ursolic acid derivatives. J. Nat. Prod., 2000, 63, 1619-22.
[25] Lee, J. S.; Miyashiro, H.; Nakamura, N.; Hattori, M. Two new triterpenes from the Rhizome of Dryopteris crassirhizoma, and inhibitory activities of its constituents on human immunodeficiency virus-1 protease. Chem. Pharm. Bull., 2008, 56, 711-4.
[26] Yim, E. K.; Lee, M. J.; Lee, K. H.; Um, S. J.; Park, J. S. Antiproliferative and antiviral mechanisms of ursolic acid and dexamethasone in cervical carcinoma cell lines. Int. J. Gynecol. Cancer, 2006, 16, 2023-31.
ISSN 1666-7948
www.quimicaviva.qb.fcen.uba.arRevista QuímicaViva
Número 1, año 8, Abril 2009
quimicaviva@qb.fcen.uba.ar