Una visión cualitativa de cómo analizar los factores que afectan a los desplazamientos químicos medidos por medio de la espectroscopía de RMN.[a]
R. H. Contreras*, T. Llorente, G. Pagola, G. Bustamante, E. E. Pasqualini y J. I. Melo
Departamento de Física, Facultad de Ciencias Exactas y Naturales, Univ. de Buenos Aires, (C1428EHA) CABA
Email: contrera@df.uba.ar
Recibido el 27/11/08. Aceptado el 11/12/08.
Resumen
En este artículo se intenta mostrar cómo un conocimiento detallado del origen electrónico de los parámetros que se miden por medio de la espectroscopía de RMN puede ampliar en forma notable la utilidad de esa técnica para estudiar una amplia gama de problemas moleculares que pueden ser de mucho interés en diversas ramas de las ciencias. Los ejemplos que se muestran se refieren a los desplazamientos químicos, y se busca describirlos a través de nociones básicas evitando los aspectos formales que podrían cambiar el foco de la atención. Se resalta que, si bien los ejemplos dados se refieren a los desplazamientos químicos medidos en fase isótropa, la misma metodología se puede extender sin mayores dificultades a las constantes de acoplamiento indirecto de espín-espín. Algo análogo cabe decirse para cualquiera de esos parámetros medidos en fase anisótropa.
Qualitative approach for analyzing factors influencing NMR chemical shifts
Abstract
If a detailed knowledge of factors affecting high resolution NMR parameters is achieved, then the scope of this spectroscopy to study different types of molecular problems that are of interest to various scientific branches can be enhanced notably. In this work few simple examples of how chemical shifts can be employed to deepen the understanding of subtle aspects of the electronic molecular structure are discussed. The applied approach is based on sound theoretical grounds, and it is described avoiding formal aspects to take into account that they could be too involved for a substantial part of the Química Viva readership. It is highlighted that, although the discussed examples refer to isotropic chemical shifts, this approach can easily be extended to anisotropic aspects of chemical shifts as well as to the analysis of indirect spin-spin coupling constants.
Introducción.
El fenómeno de la Resonancia Magnética Nuclear, RMN,[[1]] se detectó por primera vez hace algo más de 60 años[[2]]. En una etapa muy temprana el análisis y la lectura de los espectros de RMN de soluciones químicas puras permitieron construir un código adecuado para la identificación de diversos grupos funcionales.[[3]] Desde entonces se ha mejorado esta técnica en forma prodigiosa y en la actualidad se la puede aplicar para estudiar una amplia gama de problemas moleculares. Se pueden citar entre otros, la deducción de la estructura de proteínas y nucleósidos [[4],[5],[6],[7]], la deducción de vías de transferencia de carga dentro de una molécula[[8]], análisis de las interacciones tanto intra- como intermolecular para varios casos,[[9]] etc.
Junto con los progresos que se ven diariamente en la descripción de diversas vías metabólicas y de transducción de señales, existe un progreso equivalente en cuanto a elucidación de las estructuras moleculares y de sus propiedades observables. En Química Biológica, hoy en día, se estudia de manera indirecta la interacción intermolecular con sofisticadas técnicas afines. Es quizás una profundización en el conocimiento de la estructura electrónica molecular lo que aclare las posibles interacciones tanto dentro de una molécula como con las que la rodean. A partir de una base física para conocer la estructura electrónica de una molécula, los investigadores en disciplinas tales como la Química Biológica, la Biología Celular y otras afines, podrían desplegar su creatividad para resolver problemas en un nivel más profundo. Quizás este breve artículo muestre en forma somera cómo la RMN junto con ciertas ideas físicas pueden contribuir a la comprensión de detalles sutiles de la estructura electrónica molecular y pueda ser el punto de partida para estimular nuevos estudios que lleven a una comprensión complementaria y más amplia que la actual en los temas mencionados arriba.
En este artículo se muestran los lineamientos básicos de un procedimiento cualitativo que permite comprender cómo afectan ciertas interacciones tanto intra- como intermoleculares a los desplazamientos químicos. Ese método se basa en emplear expresiones rigurosas en forma aproximada que se obtienen a partir de la teoría del propagador de polarización.[[10]] Para mostrar su utilidad, potencialidad y sencillez se lo emplea para analizar el efecto de sustituyente sobre el apantallamiento magnético en unas pocas moléculas pequeñas. También se analiza la validez general de un concepto que suele expresarse frecuentemente tanto en algunos tratados como en artículos científicos que consiste en la afirmación “las interacciones de transferencia de carga sobre el desplazamiento químico presentan un efecto ya sea apantallante (desapantallante) del núcleo en cuestión cuando esas interacciones arriman (retiran) carga electrónica del entorno átomo”.[b] Para darle mayor claridad al método, esta descripción se complementa con algunos cálculos basados en la teoría de la funcional de la densidad, “DFT” (Density Functional Theory),[[11],[12]] que indican cómo puede lograrse una idea cualitativa acerca de los efectos que producen ciertas interacciones intramoleculares sobre el desplazamiento químico. Para evitar que algunos de los lectores de Química Viva encuentren este tipo de análisis excesivamente matemático, se reducen al máximo sus aspectos formales y se mencionan referencias bibliográficas adecuadas en tal forma que los interesados puedan apreciar cabalmente la rigurosidad del mismo consultando fuentes originales. Además, por brevedad y facilidad de su comprensión, en este artículo ese análisis se restringe tan sólo a los núcleos de 15N y de 17O en unos pocos compuestos muy simples.
Es importante recordar unos pocos datos fundamentales antes de entrar en consideraciones más específicas:
Los únicos núcleos donde puede detectarse el fenómeno de la RMN son aquéllos que presentan un momento magnético nuclear, y por ende, un espín nuclear caracterizado por el número cuántico I distinto de cero.[[13]] No obstante, desde el punto de vista práctico no todos los núcleos magnéticos son igualmente útiles como sondas en estudios moleculares.[[14]] Para un mismo campo magnetostático externo (provisto por el espectrómetro) la sensibilidad de un isótopo para su observación por RMN depende fundamentalmente de su abundancia natural y de la magnitud de su factor magnetogírico. Actualmente la primera se puede mejorar enriqueciendo la muestra en un determinado isótopo, aunque en general se trata de un procedimiento muy costoso. La segunda se puede mejorar empleando espectrómetros con campos magnéticos más intensos. También existen métodos de transferencia de polarización que pueden mejorar la sensibilidad en distintos casos particulares.[[15]] Además de las cuestiones mencionadas cabe destacarse que no suele ser fácil detectar núcleos que tengan un momento cuadripolar eléctrico distinto de cero pues tal circunstancia hace que sus estados de espín nuclear presenten un tiempo de relajación muy corto.[c] Los núcleos con una cantidad par tanto de protones como de neutrones (llamados “núcleos par-par”) tienen I = 0. Tal es el caso, por ejemplo, del isótopo 12 del carbono, 12C, o de los isótopos 16 y 18 del oxígeno, 16O y 18O. Dentro de sus respectivas especies los isótopos 12C y 16O son, por mucho, los de mayor abundancia natural.[14] A primera vista tal circunstancia puede parecer un inconveniente para estudiar compuestos orgánicos; sin embargo tal hecho lleva a que los espectros de dichos compuestos se vean notablemente simplificados y pueda, por tanto, extraerse una notable cantidad de información acerca de sus estructuras electrónicas moleculares. Otro aspecto sumamente importante en la aplicación práctica de esta espectroscopía lo constituye el hecho que los núcleos que tienen I > ½, presentan un momento cuadripolar eléctrico, Q, distinto de cero, tal es el caso del 17O (vide supra).
A cada porción de la muestra se puede asignar una magnetización macroscópica que resulta de la suma de los efectos magnéticos de los electrones y de los núcleos. Ahora bien, la señal electrónica que se detecta para observar un espectro de RMN, proviene de los cambios que se producen en esa magnetización macroscópica como consecuencia de las transiciones inducidas por el campo (de radiofrecuencias) provisto por el espectrómetro entre los estados de espín nuclear. Se suele designar a esa componente de la magnetización macroscópica como la “magnetización macroscópica nuclear” en contraposición con la magnetización macroscópica que producen los espines electrónicos no apareados. Es de notar que la última, cuando está presente, es mucho más intensa que la primera, por lo que la RMN no es práctica para el estudio de la estructuras electrónicas de radicales libres porque la presencia de espines no apareados genera una magnetización que perturba la observación proveniente de los núcleos. Por ese motivo, aunque a veces no se lo mencione en forma explícita, en general los compuestos cuyos espectros se estudian por RMN de alta resolución tienen todos los electrones apareados, es decir, el estado electrónico fundamental de la molécula tiene una configuración de “capa cerrada”.[d]
El estudio del tensor del apantallamiento magnético nuclear, s.
Breves consideraciones generales sobre s.
Una magnitud física ampliamente conocida y utilizada en diversos estudios relacionados con la RMN, es el apantallamiento magnético nuclear de un determinado átomo.[[16]] Se trata de una magnitud de carácter tensorial de segundo rango, s. Como este tipo de magnitudes físicas puede resultar poco conocida para algunos lectores de Química Viva, se considera oportuno incluir algunos comentarios breves sobre sus características. Una magnitud de ese tipo siempre se puede representar por una matriz de 3x3[e]. La suma de los elementos que pertenecen a la diagonal principal se conoce como su “traza”, la que es invariante ante rotaciones. Un tensor de segundo rango siempre se puede descomponer en una parte simétrica y otra antisimétrica. En el caso del apantallamiento magnético nuclear la primera parte es la que define al desplazamiento químico; la segunda, contribuye al tiempo de relajación de los estados nucleares que definen el espectro de RMN. Por tal razón en este artículo sólo se tiene en cuenta la parte simétrica del tensor s aunque no se lo mencione en forma explícita.[f] Si las mediciones se realizan en fase isótropa, entonces tan sólo es dable observar el promedio de su traza y constituye la constante de apantallamento magnético nuclear, s. Esa cantidad está estrechamente relacionada con el desplazamiento químico, que corresponde al apantallamiento relativo respecto de el de un compuesto de referencia,[[17]] es decir,
Desplazamientoquímico= d = sRef - s (1)
que, al igual que el tensor s, suele expresarse en “partes por millón”, ppm
El tensor s es una propiedad asociada a cada núcleo perteneciente a una molécula y al diagonalizarlo, se determinan tres direcciones mutuamente perpendiculares entre sí, conocidas como las “direcciones principales” de ese tensor y los tres elementos que lo componen se conocen como sus “valores principales”.[g],[h] Debe quedar claro que las orientaciones de los ejes principales de cada tensor dependen de los elementos de simetría local del átomo en cuestión y no de los elementos de simetría de la molécula. Por otra parte, se ha calculado que, para un núcleo particular, las direcciones principales se afectan poco ante el cambio de un sustituyente.[[18]] Esto quiere decir que, para un análisis cualitativo de los factores que afectan a s, en ambos compuestos (sustituido y sin sustituir) se pueden considerar que los ejes principales difieren al máximo en unos pocos grados.[i]
El tensor de apantallamiento magnético nuclear consta de una parte diamagnética, positiva y por consiguiente apantallante, y de una parte paramagnética, negativa desapantallante,[j] s = sd+ sp. Es común ver que se acepta en la bibliografía[[19],13,[20],[21]] que el efecto de sustituyente sólo afecta al tensor paramagnético. Si bien es cierto que el mayor efecto lo sufre la parte paramagnética, esa afirmación no siempre es del todo correcta.
La parte paramagnética del tensor s expresado en términos del propagador de polarización
En esta subsección se presenta en forma breve cuáles son los fundamentos del método de análisis cualitativo que permite comprender algunos de los factores que afectan a la parte paramagnética del tensor de apantallamiento magnético nuclear, sp, debido a la presencia de un sustituyente. La constante de apantallamiento magnético nuclear del núcleo M se puede expresar (al igual que el tensor) como suma de su parte diamagnética y de su parte paramagnética:
(2)
Cuando se emplea el formalismo del propagador de polarización y se usa la aproximación RPA (Random Phase Approximation)[9], la parte paramagnética de la ec.(2) se puede escribir de acuerdo con la ecuación (3) [[22]]
(3)
donde K es una constante que no se describe en forma detallada pues la ec. (3) sólo se usa en este artículo como base para una descripción cualitativa, la cual es independiente de esa constante.
A continuación se presenta una descripción sintética de los distintos términos que aparecen en la ec. (3). Debe notarse que dentro de la llave se presentan los términos diagonales del tensor paramagnético caracterizados por aa que corresponden a las coordenadas cartesianas XX, YY, ZZ. El primer símbolo de suma (desde a = 1 hasta a = 3) corresponde a la traza del tensor de apantallamiento paramagnético. La segunda suma se realiza sobre duplas de orbitales moleculares, OM, conformadas por uno ocupado y uno vacante i,a y j,b, respectivamente. Esas duplas corresponden a lo que en la teoría de perturbaciones se denominan “excitaciones virtuales”.
son los elementos de la matriz correspondiente a la inversa de la matriz del propagador de polarización singulete, 1Wia,jb. Las matrices singuletes 1A y 1B se pueden escribir como:
(4)
(5)
donde
son integrales moleculares bielectrónicas.
Por otra parte,
(6)
(7)
se conocen como los “perturbadores paramagnéticos”, es decir, los elementos de matriz del hamiltoniano paramagnético entre un OM ocupado, i, y uno vacante, a. Debe notarse que
es el radio vector posición desde el centro de coordenadas hasta el electrón sobre cuyas coordenadas se realiza la integral. En cambio,
es el radio vector de ese electrón pero medido desde la posición del núcleo M.
Es importante resaltar que la ecuación (3) es invariante ante trasformaciones unitarias aplicadas a los OMs. Por consiguiente, se puede suponer que en las ecuaciones (3) a (7) los OMs están localizados, OML, donde se emplearon procesos de localización independientes tanto para los ocupados como para los vacantes. Esta observación sugiere introducir algunas aproximaciones algo drásticas pero correctas.[k] Debe recordarse que las expresiones (3) a (7) tan sólo se emplearán en forma cualitativa para tomar una visión conceptual acerca de cómo analizar algunos factores que pueden alterar a la parte paramagnética del tensor de apantallamiento magnético nuclear. Recuérdese que uno de los objetivos que se tiene en mente es comprender cómo actúa el efecto de sustituyente sobre los desplazamientos químicos d(15N) y d(17O)[l] en compuestos muy sencillos, especialmente en lo que respecta a las interacciones que implican transferencia de carga. (Los interesados en conocer aspectos de estos tipos de desplazamiento químico pueden consultar, p. ej., las referencias [[23],[24]]) En este punto debe recordarse que en la actualidad el método más comúnmente aplicado para el estudio de interacciones de transferencia de carga, es el de “orbitales naturales de enlace”, NBO, que introdujeron hace ya más de dos décadas Weinhold y sus colaboradores.[[25]] Por lo tanto, surge en forma directa suponer lo siguiente: los OMLs en que se piensa que están escritas las ecuaciones (3) a (7), frente a interacciones de transferencia de carga se comportan cualitativamente como los NBOs de Weinhold et al. Más abajo se discute en forma más rigurosa la validez de esta suposición.
Antes de analizar con más detalle las ecs. (3) a (7) deben hacerse algunas observaciones respecto a la primera de ellas: los efectos de sustituyente (o los efectos de cualquier interacción) pueden manifestarse a través de modificaciones ya sea en el propagador de polarización singulete o en los perturbadores. No obstante, debe recalcarse que algunas interacciones pueden afectar ambos tipos de términos.
Se definen como los términos “diagonales del propagador” aquéllos en los que se cumple que i = j y a = b, es decir, a aquéllos en que las dos “transiciones virtuales” son las mismas.[m] Debe resaltarse que los términos “diagonales” del propagador son más importantes que los no diagonales, pero para tener una participación importante en el tensor paramagnético, los respectivos “perturbadores” que intervienen en el mismo término, no deben ser “muy pequeños”.[n] Luego, es importante tener una idea de cuándo un “perturbador” lleva a una contribución significativa. Antes de discutir este punto con cierto detalle, debe mencionarse que las matrices 1A y 1B aparecen sumadas y que sólo los términos diagonales de esa suma contienen la diferencia de las energías orbitales D = ea - ei que, de ahora en adelante, se denomina “gap vacante-ocupado”, o simplemente “gap”. Para esos términos se pueden despreciar las integrales bielectrónicas en comparación con ese “gap”. Luego, en forma cualitativa se puede decir que los elementos diagonales de W son, en forma aproximada, inversamente proporcionales al “gap”. Aceptando las aproximaciones mencionadas arriba acerca del comportamiento de los OMLs, es fácil comprender cómo afectan las interacciones de transferencia de carga a los términos diagonales del propagador. Efectivamente, toda interacción que transfiere carga del orbital ocupado, i lo “empuja” hacia energías más profundas, con lo cual el “gap” se hace más grande y el término diagonal de W se hace más pequeño. Algo similar cabe decirse acerca de interacciones que arriman carga al OML vacante a: una interacción de transferencia de carga lo “empuja hacia arriba”, es decir, también produce una disminución del término diagonal de W.[[26]] Nótese que esto equivale a decir que tanto las interacciones que arriman o que alejan carga de las vecindades del núcleo cuyo tensor de apantallamiento magnético se está estudiando, pueden llevar a un efecto ya sea de apantallado o de desapantallado, dependiendo del caso particular que se trate, es decir, de cómo esas interacciones afectan a dicho “gap”.
Con respecto a los “perturbadores” de la ec. (6) pueden formularse las siguientes apreciaciones: debe recordarse que la expresión dentro del paréntesis corresponde al “operador rotación de 90º” alrededor del eje a. Por consiguiente, ese perturbador corresponde a la integral de solapamiento entre un OML ocupado rotado 90º alrededor del eje a y un OML vacante. Es decir, en muchas circunstancias es sumamente sencillo apreciar cuándo un perturbador es suficientemente grande como para que el término diagonal respectivo de la ec. (3) tenga un valor significativo.
Con respecto al perturbador de la ec. (7) debe observarse que, si en el mismo se toma como centro de coordenadas el núcleo M, entonces en el numerador aparece el mismo operador rotación que el considerado en la ecuación (6) y caben comentarios análogos acerca de cuándo ese perturbador tiene un valor significativo.[o] Por otra parte, en el denominador aparece el módulo al cubo de la distancia electrón-núcleo. Por consiguiente, si los electrones del OML sufren una interacción repulsiva (ejemplo: compresión estérica) entonces en promedio los electrones están más cerca del núcleo, con lo que el denominador del término paramagnético se hace más pequeño, y el término paramagnético se agranda, produciendo un desapantallado en el núcleo en cuestión. En un trabajo anterior se ha demostrado experimentalmente ese efecto que se produce por la proximidad de un grupo metilo sobre un átomo de flúor;[[27]] también se conocen efectos similares para el oxígeno.[[28]]
Algunas consideraciones acerca del cálculo teórico del tensor de apantallamiento magnético nuclear.
Tal como surge de la ec.(2), el tensor s puede descomponerse en una parte diamagnética y una paramagnética. Por otra parte se conocen muy bien los problemas asociados a la elección de la medida electromagnética (“gauge”) para describir tanto el campo magnetostático del espectrómetro como el de los núcleos magnéticos presentes en la molécula. Una de las formas frecuentes de superar este problema es emplear en el cálculo de una base de orbitales GIAO (Gauge̵Included Atomic Orbitals).[[29]] Un costo importante que debe pagarse por esta ventaja, es que con ese método no se pueden calcular por separado las partes diamagnética y paramagnética, tan sólo se obtiene el tensor total. No obstante, si se acepta la suposición arriba mencionada, al estudiar los efectos de sustituyente, se los puede atribuir, al menos en gran medida, a la parte paramagnética.
Algunas consideraciones teóricoconceptuales sobre el tensor de apantallamiento.
La constante de apantallamiento nuclear y la carga sobre ese átomo.
Ya se comentó en la sección 2.b que la ec. (3) y siguientes indican que las interacciones de transferencia de carga ya sea hacia las vecindades o desde las vecindades de un átomo, pueden llevar tanto a un efecto apantallante como a uno desapantallante del núcleo de dicho átomo. Una forma muy sencilla de corroborar esa predicción hecha a partir del modelo cualitativo presentado en este artículo, consiste en estudiar algunos casos sencillos para constatar si existe alguna correlación entre la carga asociada a un átomo y su constante de apantallamiento nuclear. Como primer ejemplo se calcula el valor isótropo de s(15N), es decir, s(15N), en un conjunto de aminas que pueden considerarse como moléculas de amoníaco sustituidas. Esos cálculos se realizaron con la suite de programas incluidos en el Gaussian 98,[[30]] empleando el nivel teórico dado por DFT-B3LYP/6-311G** para obtener tanto las estructuras geométricas de equilibrio como para el cálculo de la constante de apantallamiento magnético nuclear s.
En la Tabla 1 se muestran los apantallamientos s junto con los valores calculados para la “carga atómica natural” [24] en la posición del nitrógeno. En la misma Tabla 1 se indican las diferencias entre las respectivas propiedades en los dos compuestos indicados entre paréntesis empleando la numeración dada en la primera columna. Así, por ejemplo, el núcleo de N en la metilamina, según este cálculo, está desapantallado 27,7 ppm respecto a ese núcleo en el amoníaco.[p] Ya que la carga electrónica en la posición del átomo N para el amoníaco es mayor en ‑0,191 u.a. que la carga atómica del N en la metilamina, en este caso se cumple la expresión mencionada arriba referida a que todo proceso que acerca carga electrónica al N le produce un apantallamiento. Sin embargo, los casos siguientes mostrados en la misma la tabla, muestran que tal afirmación no es de carácter general. Por otra parte, en la próxima sección se discute cuál es el comportamiento del “gap” respectivo al reemplazar un protón del amoníaco por un grupo metilo. Es interesante observar que al reemplazar en la metilamina el grupo metilo por un etilo, se produce un desapantallado de 20,4 ppm, y que al reemplazar en la dimetilamina los dos grupos metilo por dos grupos etilo, se observa un desapantallado de 40,0 ppm, es decir, se observa que se cumple en forma aproximada la ley de la aditividad de efectos. En general se notan discrepancias grandes entre los valores medidos y los calculados. Según se observa en los datos experimentales que se presentan en la Ref. [22] los valores del apantallamiento magnético nuclear del 15N en general dependen en forma bastante notable del solvente con que se preparó la muestra. Parecería observarse que en el caso del amoníaco los valores son aún más dependientes del solvente (es decir, el mismo soluto), probablemente debido a la existencia de asociaciones moleculares.
Tabla 1: constantes de apantallamiento magnético nuclear s (en ppm) del N en un conjunto de aminas. En cada caso se indica también la carga atómica natural calculada con el método NBO sobre el átomo de nitrógeno. Los valores experimentales se toman con referencia al nitrometano.
Amina
s
Exp.a
Q(N)(u.a.)
1
NH3
271,6
381.93b
-1,009
2
NH2Me
243,9
378.73b
-0,818
Dif. (1-2)
27,7
-0,191.
3
NH2Et
223,5
ca. 355
-0,832
Dif. (2-3)
20,4
21 a 30
+ 0,014
4
NHMe2
227,1
371 a 374
-0,647
Dif. (2-4)
16,8
5 a 9
-0,171
5
NHEt2
187,1
332 a 334
-0,672
Dif.(4-5)
40,0
39 a 40
+0,025
a Valores experimentales tomados de las Refs. [22].
b Mediciones efectuadas en el líquido puro (soluto y solvente son la misma sustancia).
En la Tabla 2 se muestran los resultados obtenidos para la constante de apantallamiento magnético nuclear, s, de 17O en un conjunto de cuatro alkiléteres. Allí se muestran las “cargas atómicas naturales” sobre el átomo de O, calculadas con el método NBO. Se observa nuevamente que esa carga atómica puede aumentar aunque se produzca un desapantallamiento del núcleo de oxígeno. También se observa que el reemplazo de un grupo metilo por un grupo etilo (el metilo etílico está en posición b respecto del oxígeno), lleva a un efecto de desapantallado cercano a las 29 ppm, tal como se mostró oportunamente, en forma experimental, para átomos de oxígeno di-coordinados unidos a los más variados sustratos.[[31]]
Tabla 2: Cálculo de las constantes de apantallamiento magnético nuclear s (en ppm) de 17O en un conjunto de cuatro alkiléteres. También se muestran los valores calculados para la carga atómica natural (en u.a.) en la posición del oxígeno. Los valores experimentales se toman con referencia al agua. Los valores experimentales están referidos al apantallamiento del agua.
s(17O)
Exp.a
Nat. (O)q
1
Dimetileter
321,5
52,5
-0,553
2
Etilmetileter
293,7
22.5
-0,572
Dif. (1 – 2)
27,8
30,0
+0,019
3
Etilmetileter
293,7
22.5
-0,572
4
Dietileter
264,5
-6,5
-0,591
Dif. (3 – 4)
29,2
29.0
+0,019
a Valores experimentales tomados las Refs. [23]
Cómo aprovechar más ampliamente un cálculo del tensor de apantallamiento magnético Nuclear. (Análisis de los autovectores y autovalores de s(15N) y de los “gap” correspondientes).
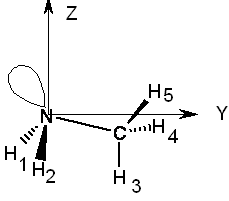
En la Tabla 3 se muestran los autovalores calculados para los tensores de apantallamiento del 15N para el NH3 y para la NH2CH3. En la misma Tabla se muestran los esquemas de las moléculas con los ejes principales de dichos tensores. Se sabe que la primera molécula (amoníaco) presenta un eje de simetría triple que se indica con Z en el esquema allí inserto. Para un átomo dentro de una molécula que tiene una ubicación tal que presente este tipo de simetría, los ejes principales están determinados por el eje de simetría triple y por otras dos direcciones cualesquiera que sean perpendiculares entre sí y perpendiculares a dicho eje de simetría.[q] Como puede advertirse, esa situación se cumple en los datos mostrados en la Tabla 3, donde además se observa que los dos autovalores iguales (XX e YY, degenerados) son más grandes que el autovalor ZZ. Esto quiere decir que la contribución paramagnética ZZ tiene mayor valor absoluto que las paramagnéticas correspondientes, XX e YY. Para comprender más profundamente el problema, en la misma Tabla 3 se indican los valores principales calculados en la metilamina, NH2Me empleando el mismo nivel de teoría que el usado en el cálculo de s en el amoníaco.[r] Debe notarse que la metilamina tiene un plano de simetría que es bisector del ángulo formado por los dos enlaces N-H, y contiene tanto al enlace N-C como al C-H3 que se ubica en forma antiperiplanar con el par no ligante del nitrógeno (efecto “Perlin”). El autovector Y no coincide exactamente con la dirección del enlace N-C sino que forma un ángulo de unos pocos grados (este ángulo ha sido exagerado intencionalmente en el esquema).
Al observar los resultados obtenidos para el amoníaco se nota que el autovalor ZZ del tensor paramagnético es el más grande (en valor absoluto) pues el autovalor correspondiente al tensor total está desapantallado en 62,4 ppm respecto de los autovalores XX e YY. En esta forma es muy fácil ver cuáles son las contribuciones más importantes al tensor paramagnético. Primero conviene analizar el comportamiento de los perturbadores. Para ello piénsese en la rotación de un OML ocupado alrededor del eje Z: un enlace sN-H se superpone en forma significativa con un antienlace s*N-H. Si se piensa que esta dupla de OMLs ocupado-vacante se repite, se está frente a un elemento diagonal del propagador de polarización, ec. (3), y, por consiguiente, su contribución a s, es inversamente proporcional al “gap vacante-ocupado”. Debido a la simetría de la molécula, hay tres expresiones iguales de ese tipo.
Nótese que las contribuciones XX e YY deben contener como términos principales de la parte paramagnética, por ejemplo para el XX, el que se obtiene al aplicar una rotación de 90º alrededor del eje X, que lleva al par no ligante sobre el eje Y. Debido al eje de simetría triple, algo análogo vale decirse acerca del autovalor YY.
Para que el estudio sea más completo, se procede ahora a analizar en forma breve el comportamiento de las energías orbitales que definen los “gaps” relevantes tanto en el amoníaco como en la metilamina. Esas energías orbitales se calcularon con el método NBO y se muestran en la Tabla 4. Aquí es muy importante recalcar que las energías orbitales de dos sistemas independientes (por ejemplo, el amoníaco y la metilamina) no se pueden comparar entre sí pues los respectivos ceros no están definidos en forma unívoca para ambos sistemas moleculares. Para dos moléculas distintas lo que tiene sentido comparar son los “gaps” vacante-ocupado análogos. Conviene comenzar este análisis retomando las consideraciones hechas arriba acerca de los términos del propagador singulete que definen el autovalor ZZ en el amoníaco. Según se aprecia en la Tabla 4, para el amoníaco los tres términos iguales que corresponden a la rotación alrededor del eje Z de un enlace sN-H están afectado por un “gap” (3-1) = 1,0457 h, mientras que en la metilamina hay un término de ese tipo que está afectado por un “gap” (3-1) = 1,0316 h, mientras que el que corresponde al solapamiento de un enlace sN-H con el antienlace s*N-C está afectado por el “gap” (5-1) = 0.9655 h, y el que corresponde al solapamiento de sN-C con s*N-H está afectado por el “gap” (3-4) = 0,9288. Se observa que en los tres casos los “gaps” relevantes son menores en la metilamina, particularmente los que involucran ya sea al enlace sN-C o al antienlace s*N-C. Esto indica que el autovalor ZZ en la metilamina debe ser menor (tiene la componente paramagnética de mayor valor absoluto) que el autovalor ZZ en el amoníaco, tal como muestran los cálculos presentados en la Tabla 3 donde esa diferencia es de 21,2 ppm. Aunque parezca demasiado reiterativo, conviene decir una vez más, que este análisis es cualitativo, donde, por esa razón no se hicieron consideraciones acerca del diferente solapamiento de los orbitales involucrados en cada caso, o de otras diferencias que deberían tenerse en cuenta si se pretendiera obtener un resultado más cuantitativo.
Tabla 3: Valores principales (en ppm) del tensor de apantallamiento magnético nuclear en el amoníaco y en la metilamina. En los diagramas se muestran las orientaciones de los autovectores. Nótese que los ejes coordenados deben formar una terna directa. Por ese motivo, se supone que el eje X es perpendicular al papel, apuntando hacia arriba de la página.
NH3
NH2CH3
XX
YY
ZZ
XX
YY
ZZ
292,4
292,4
230,0
270,0
252,9
208,8
Ahora se pueden comparar los otros dos autovalores, XX e YY, en el amoníaco con los correspondientes en la metilamina. Obsérvese que este último compuesto no presenta un eje de simetría triple y por lo tanto, se “ha levantado” la degeneración de esos dos autovalores. Para empezar, piénsese en el autovalor XX. El término que más debe afectar este autovalor es el que corresponde a rotar el par no ligante alrededor del eje X. El “gap” correspondiente a ese término (según la Tabla 4) es (5-1) = 0,9655 h, mientras que el término que más afecta en la metilamina al autovalor YY es la rotación del par no ligante alrededor del eje Y; el “gap” relevante en este caso es (3-2) = 0,7322h es decir, es notablemente menor que el “gap” que afecta al autovalor XX. Este resultado está de acuerdo con lo que muestra la Tabla 3 donde el autovalor YY está desapantallado respecto del XX en 17,1 ppm
Tabla 4: Energías de los orbitales NBO en las moléculas de amoníaco, NH3, y metilamina, NH2CH3. Recuérdese que en la salida de este programa las energías orbitales están expresadas en unidades atómicas, es decir, hartrees, 1,0000 h = 27,212 eV = 627,51 kcal/mol = 2,6255 x 103 kJ/mol
NH3
“Gap”
NH2CH3
“Gap”
1
sN-H
- 0,6192
(3-1) = 1,0457
- 0,6004
(3-1) = 1,0316
2
LP(N)
- 0,3309
(3-2) = 0,7574
- 0,3020
(5-2) = 0,6671
3
s*N-H
+ 0,4265
--
+ 0,4312
(3-2) = 0,7322
4
sN-C
--
--
- 0,4976
(3-4) = 0,9288
5
s*N-C
--
--
+ 0,3651
(5-1) = 0.9655
--
(5-4) = 0,8657a
a Es interesante señalar que ninguno de los términos diagonales del propagador de polarización singulete que afectan a los valores principales del tensor de apantallamiento magnético del 15N incluye este gap de energía. Se lo hace figurar en esta tabla tanto para llamar la atención sobre ese hecho como por razones de completitud.
Conclusiones
Se podrían mencionar dos tipos distintos de conclusiones, por una parte, los aspectos relacionados con los detalles que pueden comprenderse sobre la estructura molecular de los compuestos simples que se estudian. Esa parte se puede dejar, en gran medida, para que el lector lo aprecie en forma directa. Tal vez el punto que vale la pena resaltar es el siguiente: el análisis como el propuesto, permite discutir ciertas características del tensor de apantallamiento las que serían válidas aunque tan solo se hayan efectuado mediciones en fase isótropa. En el otro tipo de conclusiones cabe resaltar ciertos aspectos metodológicos referidos a las aproximaciones introducidas y que, a primera vista, podría presentar algún tipo de objeciones teóricas. Tal es el caso de suponer que en varios aspectos el comportamiento de los orbitales tanto ocupados como virtuales que se encuentran a través de un desarrollo perturbativo, puede considerarse que es similar al de los orbitales NBO. En ese sentido es importante notar que los “gaps” vacante-ocupado que se determinaron para los elementos diagonales del propagador de polarización empleando las energías de los orbitales NBO, llevan a una descripción cualitativa correcta del comportamiento de los términos diagonales del tensor de apantallamiento magnético nuclear.
Agradecimientos
RHC agradece el apoyo económico dado por el CONICET (PIP 5119/059) y por UBACYT (proyecto X047).
Referencias
[a]. Este artículo se obtuvo como parte de los trabajos prácticos correspondientes al curso para graduados titulado “Física Molecular: Estructura electrónica molecular y los parámetros de RMN de alta resolución”, que RHC dictara en el Departamento de Física de la FCEN-UBA durante el segundo cuatrimestre de 2008.
[b] Esa afirmación es equivalente a decir que producen desplazamientos hacia mayor o menor campo magnético, o hacia menor o mayor frecuencia, respectivamente.
[c] Salvo si el gradiente del campo eléctrico creado por los electrones en la posición del núcleo es cero.
[d] Lo que también suele expresarse diciendo que el estado electrónico molecular fundamental corresponde a una configuración singulete.
[e] El razonamiento inverso dado por la expresión: “Toda matriz de 3x3 representa a un dado tensor” no es válido..
[f] Todo tensor simétrico con elementos reales, es diagonalizable y sus autovalores son reales.
[g] Las direcciones principales también se conocen como “eigenvectors” o “autovectores”, mientas que los valores principales también se conocen como “eigenvalues” o “autovalores”
[h] ¿Cómo se puede describir en forma sencilla la diagonalización de un tensor simétrico de segundo rango? Basta pensar en una magnitud vectorial como la velocidad, la fuerza, etc. Ese vector se puede expresar mediante tres componentes en un sistema de ejes coordenados. Si se quisiera “diagonalizar” un vector, bastaría con tomar la dirección del vector para definir uno de esos ejes, entonces basta con saber su módulo para describirlo. Podría decirse “el vector se ha diagonalizado”. El módulo es para un vector lo que la traza es para un tensor de segundo rango.
[i]. No obstante, debería tenerse mucha prudencia antes de generalizar esta afirmación.
[j]. Esta expresión debe tomarse con cuidado cuando el átomo cuyo apantallamiento nuclear se estudia es de H.
[k] Se destaca que los OML que produce el método NBO permite asociar a cada OML con funciones químicas tales como enlaces, pares no enlazantes, antienlaces y orbitales de “core” (capas internas),
[l] Si bien en este trabajo sólo se mencionan los corrimientos químicos de esos dos núcleos, estas ideas se pueden aplicar a cualquier otro que sea activo en RMN.
[m] Se recomienda no confundir los “términos diagonales” de 1W con los elementos diagonales del tensor de apantallamiento magnético nuclear.
[n]. Sin lugar a dudas, “muy pequeños” es una expresión muy difusa. Aquí se la usa en este sentido: cuanto mayor es sea el perturbador, más cuidado hay que tener al analizar el término de 1W.
[o] Debe observarse que al tomar el origen de coordenadas en el núcleo, se está menospreciando el problema de la dependencia de los resultados con la medida electromagnética que se emplea para describir tanto el campo magnético del espectrómetro como del producido por los núcleos magnéticos. No obstante, no debe olvidarse que este análisis es solamente cualitativo.
[p] Recuérdese que puede expresarse lo mismo diciendo que el 15N de la metilamina tiene un desplazamiento químico mayor que el del amoníaco en 27,7 ppm.
[q] Esta propiedad no es privativa del tensor s. Téngase presente que lo mismo sucede con, por ejemplo, el tensor de inercia de un cuerpo que presenta un eje de simetría triple.
[r] Respecto de la salida del programa Gaussian 98, debe decirse que, para la metilamina, se ubicaron los ejes y se designaron en forma adecuada para que dichos ejes coordenados sean los mismos en ambos diagramas.
[1]. I. P. Gerothanassis, A. Troganis, V. Exarchou and K. Barbarossou. Nuclear Magnetic Resonance (NMR) Spectroscopy: Basic Principles and Phenomena, and their Applications to Chemistry, Biology and Medicine. Chemistry Education: Research and Practice in Europe. (2002) Vol. 3, Nº 2, pp. 229-252.
[2]. a) F. Bloch, W. W. Hansen y M. Packard, Phys. Rev., 69, 127 (1946); b) E. M. Purcell, H. C. Torrey y R. V. Pound, Phys. Rev., 69, 37 (1946).
[3]. R. T. Morrison y R. N. Boyd. Organic Chemistry. Prentice Hall International, Inc. (1992).
[4]. K. Wurhrich, Science, 243, 45 (1989)
[5]. D. A. Case, Curr. Opin. Struct. Biol. 10,197 (2000).
[6]. J. A. Vila, J. M. Aramini, P. Rossi, A. Kuzin, M. Su, J. Seetharaman, R. Xiao, L. Tong, G. T. Montelione, H. A. Scheraga, Proc. Nat. Acad. Sci. USA, 105, 14389 (2008).
[7]. G. Bifulco, P. Dambruoso, L.i Gomez-Paloma y R. Riccio, Chem. Rev., 107, 3744, (2007).
[9]. R. H. Contreras y J. E. Peralta, Prog. NMR Spectrosc. 37, 321 (2000).
[10]. J. J. Oddershede, en: Advances in Quantum Chemistry, Vol.11, ed. P.-O- Löwdin, Academic Press, Nueva York, 1978, p.275.
[11]. R. G. Par y W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, Nueva York, 1989.
[12]. N. H. March, Electron Density Theory of Atoms and Molecules, Academic Press, San Diego, Californ ia, USA, 1992.
[13]. H. Kopfermann, Nuclear Moments, Academic Press, New York, (1958).
[14]. J. W. Emsley, J. Feeney y L. H. Sutcliffe, High Resolution Nuclear Magnetic Resonance Spectroscopy, Pergamon Press, Oxford, 1965.
[15]. J. Mason (ed), Multinuclear NMR, Plenum Press, Nueva York, 1987.
[16]. T. Helgaker, M. Jaszuński y K. Ruud, Chem Rev. 99, 293 (1999).
[17]. R. K. Harris, E. D. Becker, S. M, Cabral de Menezes, P. Granger, R. E. Hoffman y K. W. Zilm, Pure Appl. Chem., 80, 59 (2008).
[18]. A. M. Orendt, R. R. Biekofsky, A. B. Pomilio, R. H. Contreras y J. C. Facelli, J. Phys. Chem. 95, 6179 (1991)
[19]. A. Saika y C.P.Slichter, J. Chem Phys., 22, 26, (1954).
[20]. W. J. Hehre, R. W. Taft y R. D. Topsom, Prog. Phys. Org. Chem., 12, 159 (1976).
[21]. W. Adcock, D. Lünsmann, J. E. Peralta y R. H. Contreras, Magn. Reson. Chem., 37, 167 (1999).
[22]. R. H. Contreras, M. C. Ruiz de Azúa, C. G. Giribet, M. B. Ferraro, A. C. Diz, Análisis Químico-Cuántico de los parámetros de resonancia magnética nuclear, en Nuevas Tendencias, Química Teórica, S. Fraga, Editor. Consejo Superior de Investigaciones Científicas, Vol. II, Cap. VI, Madrid, 1989; c) R. H. Contreras, C. G. Giribet, M. C. Ruiz de Azúa, M. B. Ferraro, Electronic Origin of NMR Parameters en Studies in Physical and Theoretical Chemistry, S. Fraga, Editor, Vol. 77(B), p. 212, Elsevier, Amsterdam, 1992.
[23]. a) M. Witanowski, L. Stefaniak y G. A. Webb, Nitrogen NMR Spectrosc. En Annual Reports on NMR Spectroscopy, Ed. G. A. Webb. Vol. 11B, Academic Press, London, 1981; b) M. Witanowski, L. Stefaniak y G. A. Webb, Nitrogen NMR Spectrosc. En Annual Reports on NMR Spectroscopy, Ed. G. A. Webb. Vol. 18, Academic Press, London, 1986.
[23]. M. Witanowski, L. Stefaniak y G. A. Webb, Nitrogen NMR Spectrosc. En Annual Reports on NMR Spectroscopy, Ed. G. A. Webb. Vol. 18, Academic Press, London, 1986.
[24]. D. W. Boykin, Ed. 17O NMR Spectroscopy in Oranic Chemistry, CRC Press, Boca Raton, 1990.
[25]. A. E. Reed, L. A. Curtiss y F. Weinhold, Chem. Rev., 88, 899 (1998).
[26]. M. J. S. Dewar y R. C. Dougherty, The PMO Theory of Organic Chemistry, Plenum Press, Nueva York, 1975.
[27]. G. W. Gribble, D. J. Keavy, E. R. Olson, I. D. Rae, A. Staffa, T. E. Herr, M. B. Ferraro y R. H. Contreras, Magn. Reson. Chem. 29, 422 (1991).
[28]. D. W. Boykin, R. L. Hertzler, J. K. Delphon y E. J. Eisenbraun, J. Org. Chem., 54, 1418 (1989).
[29]. a) R. Ditchfield, Mol. Phys. 27, 789 (1974); b) J. L. Dodds, R. McWeeney y A. J. Sadlej, Mol. Phys., 41, 1419 (1980); c) K. Wolinski, J. F. Hilton y P. Pulay, J. Am. Chem. Soc., 112, 8251 (1990).
[30]. Gaussian 98, Revision A.7, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1998.
[31]. J. E. Peralta, R. H. Contreras, O. E. Taurian, F. S. Ortiz, D. G. de Kowalewski, y V. J. Kowalewski, Magn. Reson. Chem., 37, 31 (1999).
ISSN 1666-7948
www.quimicaviva.qb.fcen.uba.arRevista QuímicaViva
Número 3, año 7, diciembre 2008
quimicaviva@qb.fcen.uba.ar