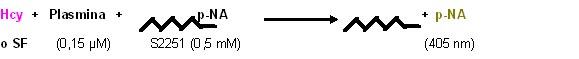Cuando se
produce injuria en un vaso sanguíneo, la pérdida de sangre se detiene por la
formación de un tapón hemostático constituido por plaquetas y una red proteica.
El fibrinógeno (Fbg: abundante proteína en la circulación) se polimeriza por
activación de los factores plasmáticos de la coagulación, formando una red
denominada fibrina. Mientras se produce la reconstrucción del vaso dañado, el
coágulo se disuelve por la acción del sistema fibrinolítico. La coagulación (formación
de la red de fibrina) y la fibrinolisis (su disolución) deben ser comprendidas
como un sistema dinámico cuyo fin es mantener una adecuada circulación
sanguínea. El desequilibrio entre estos mecanismos provoca falta o exceso de
coagulabilidad en el individuo.
Los vasos sanguíneos son
conductos de diferente calibre por donde circula la sangre; pueden
dilatarse o contraerse para regular el caudal necesario para cada condición o
actividad. Su pared, la pared vascular, está formada por una serie de capas
elásticas constituídas principalmente por colágeno, elastina y proteoglicanos,
que le otorgan resistencia y contractilidad a los vasos. El interior de los
vasos está tapizado por una única capa de células llamadas endoteliales, que
participan activamente en conservar la luz del vaso, mediar el pasaje de
nutrientes y otros solutos a los tejidos, sintetizar sustancias vasoactivas y
muchas otras que van a participar en la coagulación y fibrinolisis. En
condiciones normales el endotelio no activa la coagulación ni a las plaquetas
para generar un trombo. Dicho de otro modo, el endotelio vascular es una
superficie no trombogénica.
Las
proteínas que constituyen el sistema de coagulación se llaman factores y
generalmente se encuentran en circulación en forma de precursores enzimáticos
inactivos. Mac Farlane y Davie propusieron un sistema de reacciones
consecutivas, donde una enzima activa a su sustrato, el cual se convierte a su
vez en una enzima activa para el próximo sustrato, desencadenando un sistema
de activación secuencial (1) que conduce a la formación de trombina, la
principal enzima del sistema.
La trombina realiza múltiples funciones. Entre ellas, corta
específicamente cuatro uniones aminoacídicas de la molécula de Fbg liberando
péptidos denominados fibrinopéptidos A y B y generando monómeros de fibrina
que se polimerizan dando origen a la red de fibrina soluble. La trombina,
además, activa al factor XIII. El factor XIII activado produce uniones
covalentes entre las fibras de fibrina soluble generando fibrina estable (2).
La fibrina es degradada por la plasmina, enzima que deriva de la
activación del plasminógeno por sus activadores: activador tisular del
plasminógeno (t-PA) y activador del plasminógeno tipo uroquinasa (u-PA).
La homocisteína (Hcy) es un aminoácido que contiene un grupo sulfhidrilo
en su molécula. No es constituyente natural de las proteínas sino que es un
producto intermedio en el metabolismo de la metionina. Su concentración en la
circulación es muy pequeña: el límite superior de homocisteinemia en
individuos sanos, con niveles vitamínicos óptimos es de 12 µM (3-4). Existen
muchos factores adquiridos que regulan los niveles de Hcy. La dieta es
fundamental: el déficit de ácido fólico, vitaminas B12, B6
y/o B2 conduciría/n a niveles plasmáticos elevados del
aminoácido, un estado denominado de hiperhomocisteinemia (HHcy). El consumo
abundante de alimentos de origen animal debería estar acompañado por
cantidades adecuadas de frutas y vegetales frescos que aporten el nivel de
ácido fólico suficiente para neutralizar
la Hcy
generada.
La Hcy puede formar compuestos disulfuro con moléculas que
contienen grupos tioles, como el aminoácido cisteína o proteínas que tengan
libre el sulfhidrilo de los residuos cisteína (5).
Los niveles
plasmáticos elevados de Hcy (hiperhomocisteinemia: HHcy), están asociados a
muchas alteraciones: neurológicas (6-7), complicaciones obstétricas (8-9),
efectos teratogénicos (10), enfermedad aterotrombótica (11-12). Se ha
encontrado asociación positiva entre niveles elevados de Hcy y las distintas
formas de la enfermedad arterial: infarto de miocardio (13-14), accidente
cerebrovascular (15), y venosa: trombosis venosa profunda (16),
tromboembolismo pulmonar (17) o venoso (18-19), de modo que
la Hcy elevada constituye un importante factor de riesgo
independiente para la enfermedad aterotrombótica, afectando al sistema
vascular coronario, cerebral y periférico (20-23). La magnitud del riesgo es
similar a la de otros factores tales como colesterol elevado, tabaquismo,
hipertensión y diabetes que, a su vez, ejercen un efecto multiplicativo cuando
se combinan con hiperhomocisteinemia.
Entre los efectos nocivos de
la Hcy se ha demostrado la generación de especies oxigenadas,
altamente reactivas (peróxido de hidrógeno, radicales hidroxilo y superóxido),
que inducirían injuria en el endotelio vascular (24-25). Se ha sugerido (26)
que
la HHcy
afectaría la vasodilatación del endotelio (por disminución de la
biodisponibilidad del óxido nítrico) y las propiedades elásticas de la pared
arterial (27) (por el aumento en la síntesis y entrecruzamiento de las fibras
de colágeno). Los resultados del efecto de la HHcy sobre las plaquetas y la
actividad de los factores de coagulación y el sistema fibrinolítico son
contradictorios. Se evidencia un estado hipercoagulable en los pacientes
homocistinúricos. Mientras algunos estudios no pudieron vincular HHcy con
alteraciones de la actividad de los factores de coagulación (28) ni de la
actividad fibrinolítica (29), recientemente se ha informado (30) que
la HHcy incrementó la velocidad de coagulación, y
disminuyó la actividad fibrinolítica.
A pesar de la extensa bibliografía que apoya la
asociación entre HHcy y enfermedad vascular oclusiva, no se ha llegado aún a
un consenso sobre los principales mecanismos responsables de esta interacción.
La mayoría de los estudios publicados han evaluado la acción de
la Hcy en los procesos previos a la generación del trombo; en
cambio, en el presente trabajo se ha investigado la estructura de las redes de
fibrina y el proceso de lisis posterior.
Para contribuir a dilucidar los mecanismos involucrados
en la asociación de los niveles elevados de Homocisteína con el riesgo de
enfermedad vascular oclusiva, se evaluó el efecto in vitro del
aminoácido sobre la estructura y lisis de la fibrina. Además se estudió el
efecto de
la Hcy sobre
componentes del proceso fibrinolítico: plasmina, plasminógeno y activadores
del plasminógeno.
Materiales y Métodos:
Obtención
de fibrina
La acción de la trombina sobre el fibrinógeno en solución
produce una serie de eventos que culminan con la formación de un gel, cuyo
componente sólido (esqueleto) es la fibrina. En los distintos estudios se
utilizó una mezcla de plasmas citratados libres de plaquetas,
provenientes de individuos normales (PN*).
Alícuotas de PN*, se incubaron 1 hora, a 37ºC con soluciones de la
mezcla racémica (DL)-Hcy a diferentes concentraciones. La muestra control se
obtuvo reemplazando la solución de Hcy por solución fisiológica (SF). Se
utilizó trombina (Trb) y CaCl2 para coagular una alícuota de la
mezcla plasmática preincubada con Hcy o SF.
Los efectos de
la Hcy sobre la fibrina fueron evaluados mediante microscopía
electrónica y estudios líticos.
Estudio de las redes de fibrina por microscopía
electrónica
Modificaciones de la estructura de la red de fibrina pueden conducir a
fibrinas muy débiles e ineficientes o, por el contrario, a densas tramas
difíciles de lisar. Con el fin de esclarecer si la Hcy produce algún efecto
sobre la estructura de la fibrina, se obtuvieron geles de fibrina plasmática
obtenidos a partir de mezcla de plasmas princubados con el aminoácido y
posteriormente estabilizados. Las muestras se fijaron con glutaraldehído
(30%)- paraformaldehído (2,5%) durante 2 horas; se lavaron con buffer
cacodilato y fueron postfijadas con tetróxido de osmio (1%). Luego fueron
deshidratadas con soluciones de acetona en agua en concentraciones crecientes
(50, 70, 85 y 100 %). Las muestras se secaron por punto crítico (Baltec CPD
030, Balzers, Alemania) y fueron metalizadas para ser observadas en un
microscopio electrónico de barrido (Carl Zeizz DMS 940 A, Oberkochen, Alemania)
(31). Las observaciones se realizaron a 5 kV y se obtuvieron las fotografías
correspondientes para cuantificar las dimensiones de las fibras. Las
mediciones sobre las fotografías de las observaciones microscópicas se
realizaron con el programa Image J. Se evaluaron al menos 10 campos de igual
área, aleatoriamente seleccionados y se caracterizó cada red midiendo: número
de fibras por campo, % de red (relación porcentual entre la superficie
ocupadas por las fibras respecto al área total del campo), ancho y largo de
las fibras.
Para estudiar la acción de
la Hcy sobre la actividad de componentes del sistema
fibrinolítico, se utilizó un método cinético que evaluó el efecto
dosis-respuesta a distintas concentraciones de Hcy sobre la actividad de
plasmina y la activación del Plg.
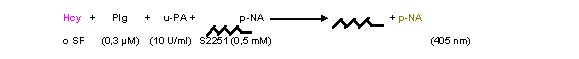
Evaluación de la lisis de fibrina
Para evaluar la lisis se utilizó un sistema en dos
etapas, separando la formación de la fibrina de su lisis. Si
la Hcy se coloca antes de formarse la fibrina se estará
evaluando la lisabilidad, como propiedad asociada a la estructura de la
fibrina. Si se coloca
la Hcy junto
con el activador del Plg al analizar la lisis de redes iguales, se evalúa
exclusivamente el efecto del aminoácido sobre el sistema fibrinolítico.
Se formaron geles de fibrina plasmática utilizando la
mezcla plasmática preincubada en presencia de distintas concentraciones de Hcy
(c.f. = 500 y 100 µM) con Trb (c.f.: 0,25 UI/ml) y CaCl2 (c.f.:
33 mM). Sobre los geles estabilizados se agregó
una solución de u-PA (25 UI/pocillo) o t-PA (10 UI/pocillo) en un volumen de
70 µl. Se monitoreó
la DO a 405
nm en función del tiempo.
Resultados
Efecto de
Hcy sobre la estructura de las redes de fibrina
En
la Figura 1 se muestran las imágenes de las
redes de fibrina obtenidas con 3.000 aumentos a partir de plasma preincubado
con Hcy 500 µM y las correspondientes controles. La caracterización de las
mismas Figura en
la Tabla 1.
|
 |
|
Figura 1: Redes de fibrina plasmática
observadas por microscopía electrónica de barrido: A: SF, B: Hcy 500 µM.
|
Tabla 1:
Caracterización de las redes de fibrina por
microscopía electrónica.
|
|
N |
Control
|
Hcy 500 µM
|
p |
|
Nº Fib/campo |
10 |
44 ± 5 |
52 ± 6 |
0,008 |
|
% Red |
10 |
67 ± 10 |
79 ± 8 |
0,001 |
|
Largo (µm) |
250 |
1,04 ± 0,25 |
0,68 ± 0,17 |
0,001 |
|
Ancho (µm) |
250 |
0,17 ± 0,04 |
0,29 ± 0,10 |
0,001 |
Resultados
expresados como promedio y desvío estándar.
El análisis estadística de los resultados se realizó por el método
de Mann Witney – Wilcoxon (para
Nº Fib/campo
y
% Red) y
el test de Student (para largo y ancho de las fibras).
Las redes de
fibrina obtenidas en presencia de Hcy 500 µM, resultaron más compactas y
ramificadas que las controles. En presencia de Hcy se formaron redes de
fibrina con mayor número de fibras/campo y que esas fibras son más gruesas y
cortas que las correspondientes a la fibrina control.
Efecto de
Hcy sobre los componentes del sistema plasminógeno-plasmina
Las redes de fibrina formadas en presencia
de Hcy mostraron diferente estructura, sugiriendo que serían más resistentes a
la lisis. Además, el aminoácido podría afectar la actividad biológica de los
componentes del sistema plasminógeno-plasmina, modificando la actividad
fibrinolítica. Con el fin de comprobar esta hipótesis, se desarrollaron
estudios cinéticos que permitieron estudiar el efecto de
la Hcy sobre:
-
la actividad
del sistema plasminógeno-plasmina,
evaluando
-
la actividad de la plasmina y
-
la activación del plasminógeno.
-
la lisis de
redes de fibrina,
estudiando
-
la lisabilidad de la fibrina
obtenida en presencia de Hcy.
-
la lisis de redes estándar de
fibrina (en ausencia de Hcy).
Acción
de Hcy sobre la actividad amidolítica de plasmina
El sustrato natural de la plasmina es la
fibrina. La plasmina corta uniones péptidicas particulares de la fibrina, pero
también puede ejercer su acción sobre fragmentos peptídicos que contengan los
sitios específicos de corte. Existen reactivos capaces de evidenciar la acción
de la enzima denominados sustratos cromogénicos y la actividad enzimática
sobre ellos se denomina actividad amidolítica. El sustrato cromogénico
contiene para-nitro anilina (p-NA), de modo que mientras que la p-NA liberada
por la acción de la plasmita es color amarillo (absorbe a 405 nm), el sustrato
cromogénico es incoloro (no absorbe a 405 nm). Por lo tanto la actividad de
la plasmina es proporcional a la intensidad del color desarrollado y puede
cuantificarse.
Se evaluó el efecto de distintas
concentraciones de Hcy (62,5; 125; 250; 500 y 1000 µM) sobre la acción
amidolítica de Plm, frente a su
sustrato cromogénico específico
S-2251 (Chromogenix, Milán Italia), en
solución reguladora a pH 7,5 determinando la liberación de p-NA (a 405 nm) en
función del tiempo, utilizando
un lector de ELISA (Reader 100, Organon Técnica, EE.UU) (32).
Cada ensayo fue realizado por triplicado.
Se caracterizaron las curvas obtenidas determinando la
pendiente y
la DO
máxima. Los resultados se muestran en
la Tabla 2.
Tabla 2:
Parámetros que caracterizan
las curvas de actividad amidolítica
de plasmina a distintas concentraciones de Hcy.
Se formaron redes de fibrina plasmática a partir de plasma (c.f. = 50 %),
con trombina bovina (c.f. = 25 U/ml), y CaCl2 (c.f. =
33 mM), de manera que todas las redes formadas en
esta experiencia fueron iguales. Sobre las redes estabilizadas se agregaron:
a)
u-PA: (50
U/pocillo) + Hcy (c.f. = 500 µM).
Control:
u-PA: (50 U/pocillo) + SF
b)
t-PA (20 U/pocillo)
y Hcy (c.f. = 500 µM).
Control:
t-PA: (20 U/pocillo) + SF
Los tiempos de lisis con (u-PA + Hcy) o (t-PA + Hcy) de fibrina
plasmática formada en ausencia de Hcy, no resultaron diferentes de sus
correspondientes controles (datos no mostrados), lo que indicaría que la
actividad de los componentes del sistema fibrinolítico no se vio afectada por
el aminoácido.
Discusión
La trombosis es considerada una patología
multicausal resultante de la interacción de factores genéticos y adquiridos.
Numerosos estudios epidemiológicos han demostrado la asociación entre los
niveles elevados de Hcy y enfermedad vascular. Actualmente, la
hiperhomocisteinemia es considerada un factor de riesgo independiente para la
enfermedad vascular oclusiva y en particular para la trombosis. Se han
realizado numerosas experiencias in vitro, ex vivo y en modelos
animales con el fin de interpretar la relación entre hiperhomocisteinemia y
enfermedad aterotrombótica, sin embargo los mecanismos involucrados no han
sido esclarecidos. La mayoría de los investigadores han estudiado la
disfunción endotelial y la actividad procoagulante entre otros efectos dañinos
de los niveles elevados de Hcy, evaluando eventos que preceden a la formación
del trombo, pero pocos (33) han investigado sobre los efectos del aminoácido
luego de iniciado el proceso de coagulación. El objetivo principal de este
trabajo fue evaluar si
la Hcy podría ser responsable de alteraciónes en la fibrina.
Por estudios de microscopía electrónica, se comprobó que
la Hcy produjo cambios en la estructura de la fibrina
plasmática,
de modo que se formaron redes compactas y ramificadas, constituidas por fibras
más cortas y gruesas que las del control.
Sauls y colaboradores (33) mostraron
imágenes obtenidas por MEB, de fibrina de plasmas de conejos inyectados
intraperitonealmente con DL-Hcy. A semejanza de nuestros resultados, estos
autores muestran que los coágulos plasmáticos de animales
hiperhomocisteinémicos parecen estar compuestos por fibras más densamente
empaquetadas.
Para dilucidar si esta estructura densa podría inducir fibrinolisis
defectuosa, se llevaron a cabo experimentos que evaluaron su lisabilidad. Por
otro lado, los niveles aumentados de Hcy podrían modificar la cinética de
generación de plasmina y/o su actividad biológica. Por lo tanto, se
evaluó además, el efecto del aminoácido sobre
componentes del sistema plasminógeno-plasmina.
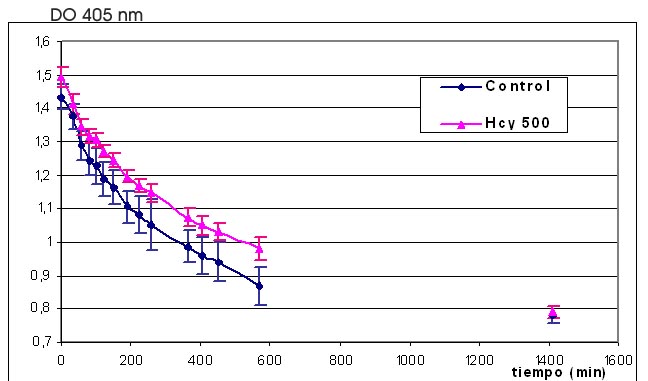
Los resultados obtenidos con u-PA y t-PA se
discutirán separadamente. A continuación se discuten las experiencias
realizadas con u-PA. Los resultados de
la Figura 2 y Tabla 4 muestran que la fibrina
asociada a Hcy resultó más difícil de lisar cuando se utilizó u-PA. Por el
contrario, cuando se lisaron geles obtenidos en “ausencia de Hcy”, con u-PA
preincubado con Hcy, se obtuvieron los mismos tiempos de lisis que el control
(u-PA preincubado con SF). Por otra parte, el aminoácido no mostró efecto en
la actividad de plasmina (Tabla 2) ni en la activación del Plg (Tabla 3)
determinadas por el método amidolítico utilizado, sugiriendo que
la Hcy no afectaría a los componentes fibrinolíticos evaluados.
Weisel y colaboradores mostraron que la velocidad de fibrinolisis está
estrechamente relacionada con el tamaño de las fibras y la arquitectura de la
red de fibrina (34). La plasmina corta transversalmente fibras individuales,
de modo que fibras gruesas son más lentamente lisadas que las finas (35). Por
otra parte, redes de fibrina ramificadas y compactas son digeridas con mayor
dificultad a causa de la elevada cantidad de fibras por unidad de volumen y la
baja difusión de componentes fibrinolíticos en esta estructura compacta (36).
Por lo tanto, considerando que
la Hcy
indujo redes de fibrina densas y ramificadas, puede postularse que la lisis
prolongada con u-PA, estaría más asociada a la estructura compacta de la
fibrina que a modificaciones en la actividad biológica de los componentes
fibrinolíticos evaluados.
Respecto a la lisis inducida por t-PA de
redes de fibrina producidas en presencia de Hcy, se observaron resultados
similares al control (Tabla 4). Según lo descripto para el u-PA, la estructura
compacta de la fibrina asociada a
la Hcy
debería haber mostrado tiempos de lisis prolongados. Sin embargo el mecanismo
de acción diferente del t-PA explicaría los resultados inesperados. La fibrina,
el principal sustrato de la plasmina, une Plg y t-PA y acelera la activación
del Plg, mientras que el u-PA no tiene afinidad por la fibrina
y activa Plg en fase fluida. La eficiencia de la fibrina como cofactor
de la activación del Plg depende de la arquitectura de las fibras: fibras
gruesas resultan mejor cofactor (37). Entonces, la estructura densa de la
fibrina asociada a Hcy sería, por un lado, más difícil de lisar, pero por otro,
las fibras más gruesas proveen más sitios de unión a t-PA, favoreciendo la
activación del Plg. Estos efectos contrapuestos explicarían que los tiempos de
lisis obtenidos con t-PA no mostraran diferencias entre las redes con Hcy y
las controles.
Estos resultados coinciden con los reportados en la bibliografía. Sauls
et al encontraron disminuida la lisabilidad con plasmina, de fibrina
obtenida a partir de Fbg aislado de plasma de conejos a los que se administró
DL-Hcy (33).
En conclusión, los tiempos de lisis de redes de fibrina obtenidos en
presencia de Hcy resultaron prolongados cuando se utilizó u-PA. Esta
alteración estaría asociada a la estructura de la fibrina, causados por
la Hcy y no a la acción del aminoácido sobre la actividad
biológica de los componentes fibrinolíticos evaluados (Plg, plasmina, u-PA y
t-PA).
Este
trabajo fue totalmente realizado in vitro para separar los diferentes
efectos que ejerce
la Hcy y poder postular mecanismos que expliquen la asociación
entre HHcy y enfermedad vascular oclusiva. Extrapolar estos resultados a lo
que podría pasar in vivo tendrían importantes consecuencias, ya que
podría inferirse que los pacientes hiperhomocisteinémicos producirían un coágulo
con una estructura más compacta y más resistente a la lisis, generando un
estado procoagulante in situ. Además, la acción de las drogas
trombolíticas estaría dificultada.
La obstrucción en la
circulación conduciría a la alteración de la reología normal y a
la secuencia de eventos que desembocarían en enfermedad aterotrombótica. Los
resultados de este trabajo contribuyen a dilucidar los mecanismos involucrados
en la asociación de la hiperhomocisteimenia y el riesgo de trombosis.
ABREVIATURAS
c.f.
Concentración final
DO
Densidad
Óptica
Fbg
Fibrinógeno
Hcy
Homocisteína
HHcy
Hiperhomocisteinemia
Plg
Plasminógeno
Plm
Plasmina
p-NA
para-nitro anilina
SF
Solución Fisiológica
t-PA
Activador tisular del Plasminógeno
Trb
Trombina
u-PA
Activador del Plasminógeno, tipo uroquinasa
Agradecimientos
Este trabajo fue financiado por la Universidad de Buenos Aires, subsidio (TX
295).
Referencias
1.
Davie E,
1995.
Biochemical and molecular aspects of the coagulation cascade.
Thomb Haemost 74:1-6
2.
Bithell TC.
Blood coagulation. En Wintrobe’s Clinical Hematology.
Tomo I.
Lea & Febiger editores, 9º edición, Londres, 1993; Pag 566-615.
3.
Ubbink J,
Becker P, Hayward Vermaak WJ, et al, 1995.
Results of B-Vitamin supplementation study used in a prediction model to
define a reference range for plasma homocysteine. Clin Chem; 41:1033-1037.
4.
Rasmussen K, Moller J, Lyngbak M, et al, 1996.
Age and gender-specific reference intervals for total homocysteine and
methylmalonic acid in plasma before and after vitamin supplementation. Clin
Chem 42:630-636.
5.
Ueland M, 1995.
Homocysteine species as components of plasma redox thiol status. Clin Chem
41:340-342.
6.
Nourhashemi F, Gillette-Guyonnet S, Andrieu S, et al,
2000.
Alzheimer disease: protective factors. Am J Clin Nutr 71:643S-649S.
7.
Regland B, Andersson M, Abrahamson L, et al, 1997.
Increased concentrations of homocysteine in the cerebrospinal fluid in
patients with fibromyalgia and chronic fatigue syndrome. Scand J Rheumatol
26:301-307.
8.
Vollset SE, Refsum H, Irgens LM, et al, 2000.
Plasma total homocysteine, pregnancy complications, and adverse pregnancy
outcomes: the Hordaland Homocysteine study. Am J Clin Nutr 71:962-968.
9.
Cotter AM, Molloy AM, Scott JM, et al,
2001. Elevated plasma homocysteine in early pregnancy: a risk factor
for the development of severe preeclampsia. Am J Obstet Gynecol
185:781-785.
10.
Mc Mullin MF, Young PB, Bailie KE, et al,
2001. Homocysteine and methylmalonic acid as indicators of folate and
vitamin B12 deficiency in pregnancy. Clin Lab Haematol
23:161-165.
11.
Van Guldener C, Stehouwer C, 2000.
Hyperhomocysteinemia, vascular pathology and endothelial dysfunction. Semin
Thromb Hemost 26:281-289.
12.
Welch GN, Loscalzo J, 1998.
Homocysteine and atherothrombosis.
N England J Med 338:1042-1050.
14.
Morris MS, Jacques PF, Rosenberg TH, et al, 2000.
Serum total homocysteine concentration is related to self-reported heart
attack or stroke history among men and women in the NHANES III. J Nutr
130:3073-3076.
15.
Miller JW, 1999.
Homocysteine and Alzheimer’s disease. Nutr Rev 57:126-129.
16.
Den Heijer M,
Blom HJ, Gerrits WB, et al, 1995.
Is
hyperhomocysteinemia a risk factor for recurrent venous thrombosis?
Lancet
345:882-885.
17.
Falcon CR, Cattaneo M, Panzeri D, et al,
1994.
High prevalence of hyperhomocyst(e)inemia in patients with juvenile venous
thrombosis. Arterioscler Thromb 14:1080-1083.
18.
Ray JG, 1998.
Meta-analysis of hyperhomocysteinemia as a risk factor for venous
thromboembolic disease. Arch Inter Med 158:2101-2116.
19.
Ridker PM, Hennekens CH, Selhub J, et al, 1997.
Interrelation of hyperhomocyst(e)inemia, factor V Leiden, and risk of future
venous thromboembolism. Circulation 95:1777-1782.
20.
Ebbesen LS, 2004.
Hyperhomocysteinemia, thrombosis and vascular biology. Cell Mol Biol
(Noisy-le-grand).
50:917-30.
21.
D’Angelo A,
Selhub J, 1997.
Homocysteine and thrombotic disease. Blood 90:1-11.
22.
Bos MJ, van Goor ML, Koudstaal PJ, et al, 2005.
Plasma homocysteine is a risk factor for recurrent vascular events in young
patients with an ischaemic stroke or TIA. J Neurol 252:332-7.
23.
Graham IM, Daly LE, Refsum HM, et al, 1997.
Plasma homocysteine as a risk factor for vascular disease: The European
Concerted Action Project.
JAMA 277:1775-1781.
24.
Misra H, 1974.
Generation of superoxide free radical during the autooxidation of thiols. J
Biol Chem 249:2151-2155.
25.
Lambert J, van der Berg, Steyn M, et al, 1999.
Familial hyperhomocysteinaemia and endothelium- dependent vasodilation and
arterial distensibility of laege arteries. Cardiovasc Res 42:743-51.
26.
Mujumdar V, Aru G, Tgagi S, 2001.
Induction of oxidative stress by homocyst(e)ine impairs endothelial function.
J Cell Biochem 82:491-500.
27.
Majors A, Ehrhart L, Pezacka E, 1997.
Homocysteine as a risk factor for vascular disease. Enhanced collagen
production and accumulation by smooth muscle cells. Arterioscler Thromb
Vasc Biol 17:2074-2081.
28.
Quintana I,
2003. Acción de la homocisteína sobre el sistema hemostático.
Tesis doctoral. Facultad de Ciencias Exactas y Naturales. Universidad de
Buenos Aires.
29.
Sabovic M, Blinc A, 2000.
Biochemical and biophysical conditions for blood clot lysis. Eur J Physiol
440 [Suppl]: R134-R136.
30.
Chia S, Wilson R, Ludlam CA, et al,
2005.
Endothelial dysfunction in patients with recent myocardial infarction and
hyperhomocysteinaemia: effects of vitamin supplementation. Clin Sci
108:65-72.
31.
Sugo T, Nakamikawa C, Yoshida N et al, 2000.
End-linked homodimers in fibrinogen Osaka VIwith a B-chain extention lead to
fragile clot structure. Blood 96:3779-85.
32.
Carr ME, Alving BM, 1995.
Effect of fibrin structure on plasmin-mediated dissolution of plasma clots.
Blood Coagul and Fibrinolysis 6:567-573.
33.
Sauls DL,
Wolberg AS, Hoffman M, 2003.
Elevated plasma homocysteine leads to alterations in fibrin clot structure and
stability: implications for the mechanism of thrombosis in
hyperhomocysteinemia. J Thromb Haemost 1:300-6.
34.
Weisel J, Veklich Y, Collet J Francis C, 1999.
Structural studies of fibrinolysis by electron and light microscopy. Thromb
Haemost 82:277-282.
35.
Collet JP, Park D, Lesty C, et al, 2000.
Influence of fibrinnetwork conformation and fibrin fiber diameter on
fibrinolysis speed. Dynamic and structural approaches by confocal microscopy.
Arterioscler Thromb Vasc Biol 20:1354-1361.
36.
Kolev K, Machovich R, 2003.
Molecular and cellular modulation of fibrinolysis. Thromb Haemost
89;610-21.
37.
Gabriel DA, Muga K, Boothroyd EM, 1992.
The effect of fibrin structure in fibrinolysis. J Biol Chem
267:24259-24263.