![]()
ISSN 1666-7948
www.quimicaviva.qb.fcen.uba.ar
Número 3, año 3, septiembre 2004
quimicaviva@qb.fcen.uba.ar
|
|
Revista QuímicaViva Número 3, año 3, septiembre 2004 quimicaviva@qb.fcen.uba.ar |
Tuberculosis: un viejo enemigo
Hector Ricardo Morbidoni*
Cátedra de Microbiología- Facultad de Ciencias
Médicas- Universidad Nacional de Rosario (UNR)- Santa Fe 3100 (2000) Rosario
Recibido 11 de agosto de 2004
Aceptado 25 de agosto de 2004
Resumen
Mycobacterium tuberculosis, agente causal de la tuberculosis humana, es responsable de casi tres millones de muertes por año en el mundo, siendo además uno de los patógenos oportunistas de mayor incidencia en pacientes HIV+. El tratamiento de la tuberculosis requiere varios antibióticos durante por lo menos seis meses, lo cual causa un elevado grado de incumplimiento. Esta situación favorece la aparición de cepas clínicas resistentes a una o más drogas. Estudios llevados a cabo durante los últimos años han identificado los blancos moleculares de las drogas corrientemente en uso y sus mecanismos de resistencia mas frecuentes. La información generada ha evidenciado un dato de gran interés: la mayoría de las drogas especificas activas contra M. tuberculosis (como Isoniacida, Etionamida, o Pirazinamida) afectan la síntesis de ácidos grasos (incluyendo los ácidos micólicos, ácidos grasos de cadena muy larga presentes en micobacterias) o de componentes de la pared celular como el caso arabinogalactano cuya síntesis es inhibida por la droga Etambutol. Estos resultados han generado un gran interés en el estudio de la síntesis de estos componentes celulares para identificar nuevos blancos aptos para el diseño de fármacos, lo cual trae esperanza para lograr mejores drogas para el tratamiento de la tuberculosis.
Palabras claves: tuberculosis, resistencia a drogas, antibióticos, mecanismo de acción, ácidos grasos.
Tuberculosis: an old enemy
Abstract
Mycobacterium tuberculosis, the causative agent of human tuberculosis, is responsible for almost three million deaths annually worldwide, being at the same time one of the prevalent pathogens affecting AIDS patients. The therapeutic treatment of tuberculosis requires the use of several anti-mycobacterial drugs for a period of six months, leading to a high level of non- compliance. This situation favors the appearance of clinical strains resistant to one or more drugs. Studies performed over the last few years have identified the molecular targets for the currently used anti-mycobacterial drugs and the most frequent mechanisms of resistance. The information generated have pointed out a very interesting fact: the majority of the specific anti-mycobacterial drugs –such as Isoniazid, Ethionamide or Pyrazinamide- affect the synthesis of fatty acids (including mycolic acids, long-chain fatty acids that are a hallmark of mycobacteria), or the synthesis of components of the cell wall such as arabino-galactan, inhibited by Ethambutol. These results have generated great interest in the study of the biosynthetic pathways of those cell components with the goal of identifying new targets suitable for the design of novel drugs, which brings renewed hope to achieve the goal of obtaining better drugs to treat this dreadful disease.
Key words: tuberculosis, drug resístance, mechanism of action, antibiotics, fatty acids.
La tuberculosis (TB), causada
por el bacilo Mycobacterium tuberculosis (MTb) es una “vieja”
enfermedad en términos de su documentacion
 histórica y es todavía un serio
problema de salud pública, no solo para países sub-desarrollados, si no
también para países industrializados que se consideraban hasta hace poco
tiempo a salvo de ella. Las cifras actuales indican que cada año hay alrededor
de 50 millones de nuevos casos de TB en el mundo, con aproximadamente tres
millones de muertos, lo que lo convierte en el agente infeccioso predominante
(1).
histórica y es todavía un serio
problema de salud pública, no solo para países sub-desarrollados, si no
también para países industrializados que se consideraban hasta hace poco
tiempo a salvo de ella. Las cifras actuales indican que cada año hay alrededor
de 50 millones de nuevos casos de TB en el mundo, con aproximadamente tres
millones de muertos, lo que lo convierte en el agente infeccioso predominante
(1).
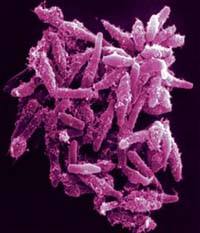
Fig. 1: Micrografia electronica de celulas de Mycobacterium tuberculosis tratadas con la droga anti-tuberculosa Isoniacida (48 h de tratamiento).
La aparición del virus de la Inmuno Deficiencia Humana (HIV) empeoró la situación descripta, debido a que los pacientes con serología positiva para HIV en las categorías A3, B3 , C1, C2 y C3 (SIDA) a menudo sufren infecciones diseminadas por MTb o por M. avium (una micobacteria patógena oportunista) constituyendo una causa frecuente de muerte. El resurgimiento de la TB en países industrializados a mediados de los años 80 demostró que, lejos de estar terminada, la batalla comenzaba nuevamente y que era necesario estudiar la fisiología de M. tuberculosis para comprender sus mecanismos de virulencia e identificar los blancos moleculares susceptibles de ser utilizados en el diseño de nuevas drogas.
Drogas anti-tuberculosas en uso: cúanto sabemos y cúanto falta por saber
Hasta el presente, las drogas empleadas para el tratamiento de la tuberculosis son limitadas, tanto en número y eficacia, como en los blancos moleculares sobre los que actúan. La larga duración del tratamiento antibacilar y la presencia de efectos colaterales determinan una mayor complejidad en la terapéutica. De hecho, el incumplimiento del tratamiento es una de las causas implicadas en la aparición de cepas multi-resistentes (definidas como resistentes a dos o más fármacos), justificando los esfuerzos en la obtención de nuevas drogas para el tratamiento de esta enfermedad (3). El régimen óptimo de tratamiento de cepas no resistentes de M. tuberculosis consiste en un cóctel de tres, cuatro o cinco agentes de primera línea, según la categorización clínica, radiológica y bacteriológica del paciente (2): Isoniacida (INH), Rifampicina (RIF), Pirazinamida (PZA), Etambutol (ETB) y Estreptomicina (SM). El tratamiento habitual consiste en una primera fase de dos meses de duración con INH, RIF, PZA y EMB seguida de una segunda fase, de cuatro meses de duración con INH y RIF. Esta combinación de tratamientos es generalmente efectiva aún si el microorganismo es resistente a una de las drogas usadas. Es interesante destacar que a excepción de RIF (un inhibidor de RNA polimerasas procarióticas) y SM (un inhibidor de síntesis de proteínas), los demás quimioterápicos usados actúan sobre la síntesis de ácidos grasos complejos de las micobacterias (4). A continuación se detallarán los mecanismos de acción de los mismos:
Rifampicina (RIF) es un antimicrobiano de amplio espectro, introducido en la terapia anti-tubercular en la década del 70. Se ha demostrado que RIF es capaz de inhibir específicamente el proceso de transcripción mediante la inhibición de la subunidad β de la RNA polimerasa (5). El hallazgo de mutaciones en el gen rpoB (que codifica la subunidad mencionada) en cepas de MTb RIFR confirmó su mecanismo de acción (6).
Estreptomicina (SM) es un producto de la bacteria Streptomyces griseus (un microorganismo emparentado con las micobacterias) con actividad sobre bacterias Gram (+) y Gram (-). Este antibiótico se une a un sitio en la subunidad 30S del ribosoma impidiendo la iniciación de la síntesis proteica. Uno de los mecanismos más frecuentes de resistencia a SM consiste en la acilación de la droga por enzimas modificadoras de aminoglucósidos, sin embargo, la resistencia a SM en micobacterias se origina en gran medida en mutaciones en la subunidad 30S, incluyendo mutaciones puntuales en el gen rpsL que codifica la proteína ribosomal S12 (7) y en el operón rrs que codifica el 16S rARN. La ausencia de mutaciones en estos genes en cepas MTb SMR indica que posiblemente haya otros mecanismos de resistencia.
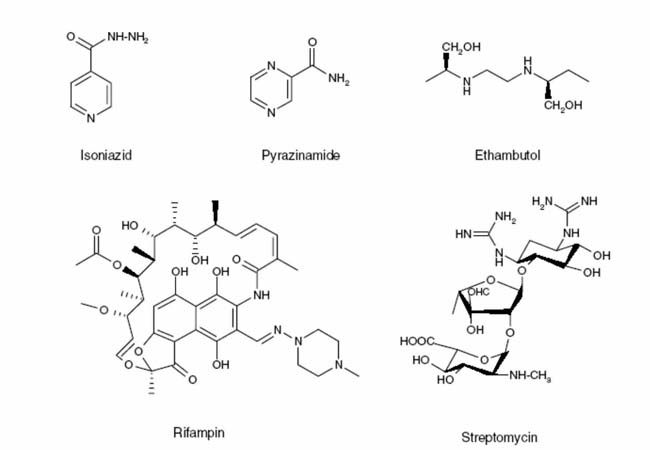
Fig. 2. Estructura de las principales drogas usadas para el tratamiento de la tuberculosis.
Pirazinamida es un derivado de la nicotinamida que se utiliza desde los años 80, generalmente asociada a RIF e INH. A pesar de su buena actividad “in vivo” en humanos y modelos animales, PZA muestra poca actividad “in vitro” a pH neutro, sin embargo esta actividad se manifiesta si el pH es ácido (5.6) sugiriendo una activación por ácidos (8). Este pH ácido es el existente dentro de los fagosomas de los macrófagos en donde usualmente reside Mtb. Interesantemente PZA es activo sobre MTb pero no sobre M. bovis ni la mayoría de las otras micobacterias, indicando la existencia de un blanco específico en el bacilo de Koch. El derivado de PZA, 5-Cl-PZA es sin embargo de rango más amplio (9). Se determinó que PZA es una pro-droga que requiere ser activada por una “pirazinamidasa” que la convierte en la molécula activa, el ácido pirazinoico (8). La identificación de mutaciones que confieren resistencia a PZA localizadas en el gen pncA, que codifican para la mencionada enzima, dieron soporte a esta hipótesis (8). El misterio de la resistencia a PZA en M. bovis fue aclarado al secuenciarse pncA en este bacilo, encontrándose mutaciones puntuales que eliminaban la actividad, es decir, M. bovis es naturalmente resistente a PZA. Aunque pcnA es la enzima activadora, el blanco molecular se determinó recientemente al hallarse que el tratamiento con PZA inhibía la síntesis de ácidos grasos de cadena larga (C16-C24). De la misma manera, la super-expresión del gen fasI que codifica para la enzima FASI causó resistencia a PZA (10). Sin embargo todavía no se han descripto mutaciones en fasI asociadas a PZAR.
Etambutol (EMB) es un inhibidor de la transferencia de los ácidos micólicos a la pared celular (11). Subsecuentemente se determinó que EMB inhibía la síntesis del arabino-galactano, un heteropolisacárido clave en la estructura de la pared celular y en donde se esterifican las moléculas de ácido micólico. Su análisis genético reveló que la resistencia a EMB se localizaba en el operón emb (compuesto por el regulador embR y las enzimas embA, emb y embC) por lo cual se postuló que los genes de este operón codificaban para arabinosil transferasas (12).
INH (hidrazida del acido isonicotínico) es una de las drogas más usadas en el tratamiento de TB. Tiene una Concentración Inhibitoria Mínima (CIM) de 0,05 µg/ml para cepas susceptibles de MTb y de otras micobacterias del complejo de M. tuberculosis pero es mucho menos eficiente sobre cepas de M. avium y otras micobacterias del complejo no tuberculosis. INH es una pro-droga que debe ser activada por una catalasa –peroxidasa codificada por el gen katG (14). KatG cataliza la oxidación de INH generando radicales reactivos, por lo tanto mutaciones en dicho gen impiden la activación y causan resistencia a INH, siendo la causa más frecuente de resistencia a esta droga (13); por lo tanto, drogas como Triclosan (TRC) que no requieren activación (Morbidoni, HR y L. Kremer, resultados no publicados) son fundamentales para evitar este mecanismo de resistencia. INH inhibe a la enzima enoil-ACP-reductasa – codificada por el gen inhA- que es parte del sistema FASII involucrado en la elongación de ácidos grasos a ácidos micólicos (14). La importancia de esta enzima como blanco de la acción de drogas se demostró al hallar que otras drogas –TRC, Etionamida (ETH), Diazaborina (DZB)- actúan mediante inhibición de InhA. El mecanismo de acción de INH es muy complejo ya que aunque claramente InhA es el blanco inhibido, otros genes (aphC, ndh) se encuentran involucrados en la resistencia (14).
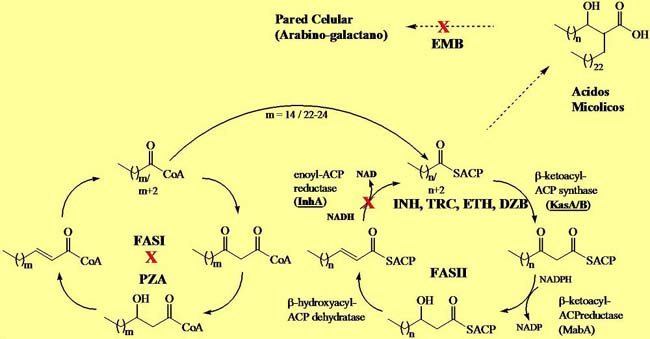
Fig. 3. Mecanismo de acción de las principales drogas anti-tuberculosas citadas en el texto. “X” denota el sitio de inhibición. PZA inhibe FASI pero no se determinó cual es la reacción inhibida.
El futuro: Las técnicas de manipulación genética han permitido identificar una serie de blancos moleculares esenciales para la viabilidad y la virulencia de M. tuberculosis. A su vez, los progresos realizados con técnicas de química combinatoria brindan la posibilidad de producir bibliotecas de compuestos que posteriormente se prueban sobre M. tuberculosis tanto sobre el crecimientos de cultivos como en infecciones de macrófagos. También se determina la actividad “in vitro” utilizando ensayos enzimáticos basados en las enzimas identificadas como esenciales (15). Lo anteriormente mencionado, en conjunto con la aplicación de programas inter-disciplinarios involucrados en la búsqueda e identificación de principios anti-tuberculosos de origen natural (presentes en plantas, microorganismos, etc) constituyen al presente, las herramientas más promisorias para la obtención de nuevas drogas que puedan utilizarse en el tratamiento de la tuberculosis.
Referencias:
1. Enarson Da, Chretien J. 1999. Epidemiology of respiratory infectious diseases. Curr. Opin. Pulm. Med., 5(3):128-135.
2. Espinal M.A. et al, 2000. Standard short-course chemotherapy for drug-resistant tuberculosis. JAMA, 283(19):2537-2545.
3. Blanchard J, 1996. Molecular mechanisms of drug resistance in Mycobacterium tuberculosis. Ann. Rev. Biochem., 65:215-239.
4. Chopra I, Brennan P, 1997. Molecular action of anti-mycobacterial agents. Tuber. Lung Dis., 78(2):89-98.
5. Levin ME, Hatfull GF., 1993. Mycobacterium smegmatis RNA polymerase: DNA supercoiling, action of rifampicin and mechanism of rifampicin resistance. Mol. Microbiol., 8(2):277-285.
6. Telenti A. et al., 1993. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet, 341(8846):647-650.
7. Finken M. et al., 1993. Molecular basis of streptomycin resistance in Mycobacterium tuberculosis: alterations of the ribosomal protein S12 gene and point mutations within a functional 16S ribosomal RNA pseudoknot. Mol. Microbiol., 9(6):1239-1246.
8. Scorpio A, Zhang Y, 1996. Mutations in pncA, a gene encoding pyrazinamidase/ nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med., 2(6):662-667.
9. Cynamon MH, Speirs RJ, Welch JT, 1998. In vitro antimycobacterial activity of 5-chloropyrazinamide. Antimicrob. Agents Chemother. 42(2):462-463.
10. Zimhony O, Cox JS, Welch JT, Vilcheze C, Jacobs WR, Jr., 2000. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat. Med. 6(9):1043-1047.
11. Takayama K, Armstrong EL, Kunigi KA, Kilburn JO, 1979. Inhibition by ethambutol of mycolic acid transfer into the cell wall of Mycobacterium smegmatis. Antimicrob. Agents Chemother., 16(2):240-242.
12. Escuyer VE et al., 2001. The role of the embA and embB gene products in the biosynthesis of the terminal hexa-arabinofuranosyl motif of Mycobacterium smegmatis arabinogalactan. J. Biol. Chem., 276(52):48854-48862.
13. Zhang Y, Heym B, Allen B,Young D, Cole S, 1992. The catalase-peroxidase gene and isoniazid resistance in Mycobacterium tuberculosis. Nature, 358 (6387):591-593.
14. Vilcheze C, Morbidoni HR, et al., 2000. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J.Bacteriol., 182(14):4059-4067.
15. Besra GS, Kremer L., 2002. Re-emergence of tuberculosis: strategies and treatment. Expert Opin. Investig. Drugs, 11(2): 153-157.
*Dr. Hector Ricardo Morbidoni
Investigador Independiente- Consejo de Investigaciones
de la Universidad Nacional de Rosario (CIUNR),
Cátedra de Microbiología- Facultad de Ciencias Médicas- UNR- Santa Fe 3100 (2000) Rosario
e-mail: morbiatny@yahoo.com
|
|
Revista QuímicaViva Número 3, año 3, septiembre 2004 quimicaviva@qb.fcen.uba.ar |