“Del vuelo
de las proteínas y de cómo lograrlo”
(Espectrometría de masa ESI)
Rosa
Erra-Balsells
CIHIDECAR-CONICET, Departamento de Química Orgánica,
Facultad de Ciencias Exactas y Naturales, UBA
Recibido 16 de agosto de 2004
Aceptado 30 de agosto de 2004
Resumen
El método “electrospray” (ESI-MS) ha extendido el uso de la La espectrometría de masa con ionización. La una gran variedad de compuestos que incluyen proteínas, glicoproteínas, oligosacáridos, nucleótidos (ADN, ARN y oligonucleótidos), fullerenos, polímeros sintéticos, compuestos inorgánicos y organometálicos entre otras especies termolábiles. Sin embargo, lo que distingue fundamentalmente a este método de ionización extremadamente suave del denominado UV-MALDI es la producción preferencial de iones gaseosos policargados y la posibilidad de su generación a partir de macromoléculas biológicas termolábiles donde la presencia de interacciones nativas no covalentes permanece inalterada. Esta caraterística es la base de su uso para estudiar desde el punto de vista estructural, cinético y termodinámico interacciones entre biomoléculas del tipo complejos proteína-proteína, complejos enzima-inhibidor, o interacciones del tipo centro activo proteico-fármaco o ADN-fármaco. El proceso de plegado y desplegado de proteínas también se puede estudiar con esta técnica. Estas aplicaciones especiales sumado al hecho de que constituye la interfase entre técnicas separativas en fase líquidas (LC, HPLC, HPCE, SEC, IEC) y la espectrometría de masa, son los factores que han contribuído al uso atractivo de la ESI-MS en áreas tan diversas como biotecnología, bioanalítica, biología molecular, química medicinal y farmacéutica y nuevos materiales incluyendo organometales y polímeros sintéticos.
Palabras
clave: ionización
"electrospray", ESI, proteómica, complejos no-covalentes; complejos
proteicos
“About how to get the
proteins to fly”
(ESI mass spectrometry)
Abstract
Electrospray ionization mass spectrometry
(ESI-MS) has made MS a viable analytical technique to study a wide range of
thermally labile compounds including proteins and glycoproteins, nucleotides
(including DNA, RNA, and oligonulcleotides), fullerenes, synthetic polymers,
inorganic and inorganic organometallic compounds. The basic difference between
soft ionization methods as UV-MALDI and ESI is that the latter provides an
inherently “gentle” ionization that has been shown to produce intact multi
charged iones from thermally labile biological molecules even biologically
relevant native noncovalent complexes. This latter capability provides the
basis for studying structurally, cinetically and thermodinamically point of
view specific biomolecular interactions, including protein-protein complex,
enzyme-inhibitor interactions, active hole protein-drug interactions and
DNA-drug interactions. Folding and unfolding protein process is also studied.
These attractive features together with the fact that ESI-MS can be hyphenated
to analytical chromatography methods dealing with liquid solutions (LC, HPLC,
HPCE, SEC, IEC, etc.) have made electrospray ionization a topic of intense
research activiy in recent years and has resulted in rapid acceptance and use
in the biotechnology, bioanalytical, molecular biology, medicinal chemistry,
pharmaceutical, new materials as organometallic and sinthetic polymers research
communities.
Key words: electrospray ionization, ESI, proteomics, non-covalent complex, protein-protein complex.
Introducción
Desde la década del 90 la espectrometría de masa (MS) se ha transformado en una técnica analítica indispensable en el campo de la química biológica. En términos generales permite hacer determinaciones estructurales, identificaciones y análisis de trazas. Es en la actualidad una alternativa muy atractiva que reemplaza a la utilísima secuenciación de Edman de polipéptidos y a métodos clásicos usados para la identificación de modificaciones post-transduccionales. Permite, además, estudiar interacciones de tipo no covalentes, por ej. estudios de la unión antígeno-anticuerpo que permiten identificar los ligandos específicos a determinados receptores. La combinación de la MS con la electroforesis bidimensional (2-D GE) y las técnicas analíticas separativas líquidas (LC, HPLC, HPEC, SEC, IEC) permite identificar proteínas. La miniaturización de la escala de trabajo al nano-litro y al nano-flujo han optimizado las características de esta técnica en lo que se refiere a tiempo y sensibilidad.
Toda esta revolución se debe a que a
fines de la década del 80 se introdujeron dos métodos suaves para la
volatilización / ionización de moleculas termolábiles. En la actualidad podemos
decir que estos métodos de ionización, ESI y UV-MALDI [1], son complementarios (Fig. 1). El primero requiere disponer del
analito en solución, en un medio polar, introducido en forma de flujo continuo
a la región donde se aplica un elevado campo eléctrico. El segundo requiere
disponer del analito sólido mezclado con un fotosensibilizador (matriz)
depositado sobre el electrodo al que se aplicará un campo eléctrico luego de
haber sido bombardeado por un láser UV [1]. Ambos son suficientemente suaves,
de manera que generan iones moleculares gaseosos intactos. El primero genera
para un dado analito de peso molecular m, una familia de iones moleculares
gaseosos policargados (m/z; m, peso molecular; z, carga; z=1, 2, 3,…,n; m/1,
m/2, …….m/n) mientras que el segundo genera preferentemente el ión molecular
gaseoso monocargado (m/z; z=1; m/1).
|
|
|
|
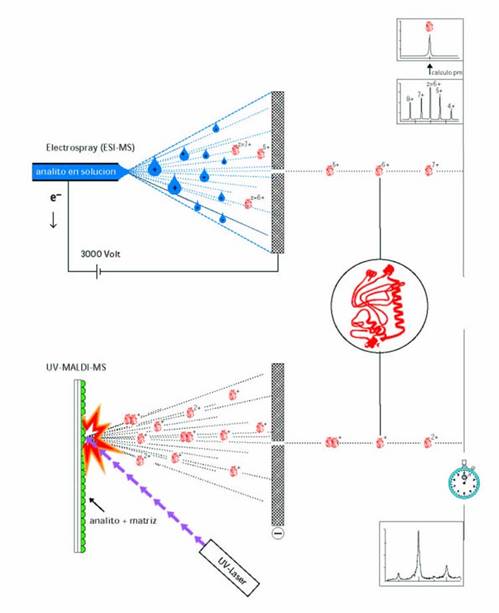
Figura1. MS de macromoléculas. En ESI-MS se generan varios
iones a partir del ión molecular (m), de diferente m/z, con 4 < z <8. A partir
de ellos se calcula el valor de m (peso molecular) (superior). En UV-MALDI se
genera el ión molecular monocargado (z=1), pequeñas proporciones del ión
doblemente cargado (m/2), y del dímero (2m/1).
El sueño se hizo realidad...”Las macromoléculas vuelan porque ESI da alas a moléculas elefánticas”
Como se detalla en el artículo
precedente [1], uno de los desafios en la década de los 80 era encontrar la
forma de analizar compuestos de alto peso molecular, termolábiles por
espectrometría de masa, y además, convertir a esta técnica en el detector de
las técnicas analíticas separativas en fase líquida (técnicas cromatográfícas
para analitos en solución líquida; LC, HPLC, HPEC, SEC, IEC) [2]. La
introducción de la técnica de ionización basada en la generación, a partir de
la solución del analito, de una nube (“spray”) formada por diminutas gotas
cargadas eléctricamente, que es sometida a la acción de una fuerte campo
eléctrico, convirtió ese ansiado sueño analítico en realidad [2]. A este método
de ionización se lo llamó “electrospray” (ES) y las diferentes mejoras
tecnológicas han conducido al método de ionización que hoy conocemos como
“electrospray ionization” (ESI) y que llevó a John B. Fenn [3] a compartir con
el Ing. Tanaka el Premio Nobel de Química 2002 [1]. Se trata del método de
volatilización / ionización de analitos más suave que se conoce a tal punto,
que se ha demostrado que material viral sometido a este método de ionización
mantiene intacta su actividad luego de ser sometido a tal proceso [4].
Como dijo Fenn: “Las
macromoléculas vuelan porque ESI da alas a moléculas elefánticas” (Fig. 2) [3].
|
|

Un poco de historia acerca del desarrollo del método de ionización ESI
A la descripción de los experimentos
iniciales efectuados por el físico John Zeleny en 1917 siguió la primera
explicación lógica del fenómeno de la generación de una nube con microgotas
cargadas efectuada por Malcolm Dole recién en 1968 [5]. Esta descripción del
principio por el que se produce el "electrospray" (ES) y su
comportamiento bajo la acción de un fuerta campo eléctrico, incluye el modelo
que contempla la formación de cargas residuales (CRM) el que ha sobrevivido
como la mejor descripción física del enigmático proceso de ioniación por
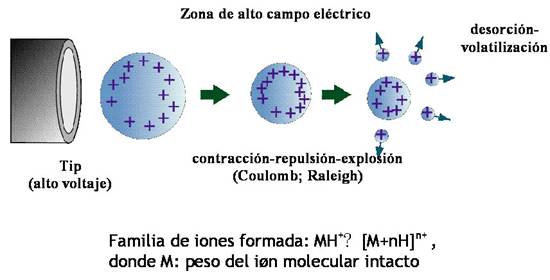
"electrospray" (ESI) (Fig. 3). De acuerdo a este modelo, las microgotas cargadas electrostáticamente
sometidas a un fuerte campo eléctrico, sufren primero una contracción de
volúmen y sus componentes pasan al estado gaseoso, cuando el número de cargas
electrostáticas del mismo signo, que se encuentran en la superficie, es mayor
que las que puede estabilizar una gota del mismo tamaño teniendo en cuenta las
repulsiones de Coulomb. En ese momento, se produce la “explosión de Raleigh”
que no es más que la eyección de moléculas de la superficie de la gota que
pasan al estado gaseoso llevándose una o varias cargas, con el objeto de
disminuir el número de cargas y las fuerzas repulsivas en la superficie. Esta
“explosión” origina a una microgota cargada de tamaño menor, la cual sigue
teniendo en su superficie un número alto de cargas electrotáticas del mismo
signo. Ésta bajo la acción del fuerte campo eléctrico vuelve a contraerse. Si
en esta gota menor no se cumple de nuevo la relación número de cargas
repulsivas a superficie total de la gota adecuada, vuelve a producirse una
“explosión de Raleigh”, con pasaje al estado gaseoso de más moléculas mono o
policargadas componentes de la gota. Esta sucesión de contracciones-explosiones
(evaporaciones superficiales) – disminución del tamaño de la gota
–contracciones- explosiones (evaporaciones superficiales) – etc., va generando
moléculas gaseosas ionizadas de los componentes de la gota orignal y por lo
tanto, de la solución de partida (Fig. 3). Las
fuerzas repulsivas que provocan estas explosiones no inducen en general la
ruptura de uniones químicas. Por lo tanto, es posible generar, a partir de
soluciones diluídas, iones moleculares gaseosos intactos de cualquier analito
presente en la solución. La particularidad de este método de volatilización /
ionización no es solamente su “suavidad” (producción del ión molecular intacto)
sino además la generación de una familia de iones debido a que el ión molecular
intacto puede alojar un número variable de cargas (ion molecular policargado;
m/z, para un dado m, z, familia de número de cargas; z= 1, 2, 3,…..n).
|
|
|
|

La mayor o menor capacidad que tendrá el analito para generar moléculas gaseosas cargadas y el número de cargas (z), depende directamente del peso molecular (m) y del carácter polar del mismo, o sea de su estructura química (tipo y número de grupos funcionales polares presentes). A mayor peso molecular y mayor número de grupos polares, mayor posibilidad de generar iones policargados (z>1) y menor posibilidad de generar iones monocargados (z=1). Además, la mayor o menor capacidad que tendrá el analito para generar iones gaseosos cargados positivamente (cationes) o negativamente (aniones) depende en primer lugar del tipo de grupos polares presentes en su estructura y en segundo lugar de las características del medio (pH, presencia de sales) y del solvente de la solución original (caracter prótico del solvente, polaridad, constante dieleléctrica, etc.).
Esta triple dependencia (estructura molecular-medio-solvente) del proceso de ionización llamado ES y su suavidad, indica por un lado el campo de aplicación del mismo el cual si bien en principio parecería limitarse al ámbito de las moléculas polares en medios polares se extiende al de las moléculas polarizables mediante modificaciones adecuadas del medio donde se las disuelve (cambio de pH, agregado de sales cuyos cationes forman complejos gaseoso estables cargados con la molécula del analito neutro, etc.).
Cabe señalar que, dependiendo de los factores antes mencionados, para un analito M su ionización en modo positivo puede deberse a la fijación de (i) n protones: [M(H)n]n+, (ii) m ó p cationes (Na+, K+, Li+, Cs+, etc.): [M(Na)m]m+ , [M(K)p]p+ , etc., (iii) la combinación de n protones y m y p cationes: [M(H)n]n+ , [M(H)n(Na)m](n+m)+ , [M(H)n(Na)m(K)p](n+m+p)+, [M(H)n(K)p](n+p)+ , [M(Na)m]m+, [M(K)p]p+ , [M(Na)m (K)p](m+p)+, etc. donde todas las combinaciones y valores de n, m y p son posibles. En modo negativo normalmente se produce la pérdida de protones [M-(H)n]n- y/o la pérdida de cationes en compuestos salinos: ej. para un compuesto M, de estructura M = N-(SO4Na)m, pueden formarse la familia de aniones {[N-(SO4Na)m]-nNa}n- con m ³ n. Esta complicación adicional en cuanto a que los iones policargados múltiples que se pueden formar dependen no sólo de la estructura del analito sino además de los componentes de la solución (pH, sales) son una fuerte desventaja del método de ionización ESI frente al método de ionización suave UV-MALDI. La pureza del analito y de la solución es crítica en el primero (Fig. 4) (Tabla 1).
|
|

Tabla 1. ESI-MS
y UV-MALDI-MS: características, utilidad, ventajas, desventajas y campo de
aplicación.
|
Método de ionización |
Límite pm teórico/Da (práctico/Da) |
Rango dinámico m/z medible teórico/Da (práctico/Da) |
Ventajas |
Desventajas |
Campo de aplicación
|
UV-MALDI
|
>300000 (~ 200000) |
23- >300000 (23-200000) |
-Ión único (m/z, m=pm; z=1)
en modos (+) y (-). -Bajo efecto de sales (~ mM). -Análisis de mezclas (ej.: huella peptídica; oligómeros). -MSn (fragmentación) estructura del compuestos: ej.
secuenciación de proteínas. -Sensibilidad: fmol-atmol. |
-Interferencia de la matriz. - No útil como detector ifenado a métodos separativos
(GC/GCL/LC/HPLC/HPCE). -Analítica s/muestra sólida. -No útil para compuestos que absorben a la lem del Laser. -Falta de reglas para selección de la matriz adecuada. |
-Péptidos -Proteínas -Glicoproteínas -Hidratos de carbono -Glicoconjugados -Nucleótidos -Nuclósidos -Esfingolípidos -Lípidos -Polímeros sintéticos polares y no polares -Moléculas polares pequeñas |
ESI
|
~ 200000 (70000) |
23~10000 (23~4000) |
-Familia de iones (m/z, m=pm; z =
1, 2, 3,..,n) en modos (+) y (-). -Suave: no rompe interacciones débiles no covalentes (ej, complejos
proteicos; organometales). -MSn (fragmentación) estructura del compuestos: ej.
secuenciaci”on de proteínas. -Sensibilidad: fmol-atmol. -Analítica sobre muestra en solución. -Gran utilidad como detector ifenado a métodos separativos (LC; HPLC;
HPCE; SEC; IEC) |
-Analitos puros. -Familia de iones moleculares policargados confunde el análisis de
mezclas. -No útil para mezclas. -Interferencia de sales. -Sólo para analitos polares y/o polarizables. -Sólo solventes polares. -Mantenimiento: frecuente por contaminaciónde la cámara de ionización. |
-Péptidos -Proteínas -Glicoproteínas -Hidratos de carbono -Glicoconjugados -Nucleótidos -Nuclósidos -Esfingolípidos -Organometales -Inorgánicos -Polímeros sintéticos polares y polarizables -Moléculas polares pequeñas. |
En los experimentos que describió Dole en 1968 [5] usó un gas inerte adicional para facilitar la desolvatación irreversible (separación irreversible de moléculas del analito y del solvente en el estado) gaseosodurante la “explosión de Raleigh”. En otros experimentos se mejoraba la eficiencia de la generación de moléculas gaseosas cargadas del analito en cuestión completamente ‘desorbidas” o libres de moléculas de solvente, calentando el conducto o aguja metálica o “tip” (Fig. 3) por el que circula la solución que va a ser volatilizada / ionizada por ES y al que se aplica un alto voltaje. Esta es la denominada “thermo spray ionization” (TES) [2,3] que es la base de la “atmospheric pressure chemical ionization” (APCI) [2].
Pero…si la técnica de ionización de moléculas por nebulización llamada ES se conocía desde la década del 60, ¿porqué recibe John B. Fenn el Premio Nobel en el año 2002? ¿Cúal ha sido su contribución?
Lo que conocemos hoy como método de volatilización / ionización ESI de real utilidad para moléculas termolábiles es una modificación del método de Dole, que Fenn presenta en un simposio en San Francisco (USA) en 1988 (Fig. 5) [6]. En dicha comunicación se describió la identificación de polipéptidos y proteínas de peso molecular 40 kDa. Fenn demostró que era posible determinar, a partir de la familia de iones generada debido a la policarga de un mismo ión molecular m, su peso molecular con una exactitud del 0,01%. Para ello, teniendo en cuenta la abundancia isotópica natural de cada elemento constituyente, se simulaba matemáticamente cúal era la mejor combinación de átomos en el ión molecular (el mejor m) que generaría la familia de iones multicargados obtenida en cada caso experimentalmente. Este resultado fue la culminación de desarrollos tecnológicos y de modelado matemático que se venían haciendo en el laboratorio de Fenn, en Yale, desde 1984 [7]. En ese año se efectuó exitosamente la utilización del "electrospray" como método de volatilización / ionización en un espectrómetro de masa. Fenn usó su conocimiento sobre el proceso denominado “free-jet expansion” en la interfase líquido-gas ionizado, y en cooperación con el especialista japonés en "jets" profesor Yamashita, modificó el arreglo experimental del ES descripto por Dole [5]. Entre otras cosas se agregó un gas inerte adicional para optimizar el proceso de desolvatación (“drying gas”), eliminando el proceso inverso o re-solvatación del ión gaseoso macromolecular formado, sin necesidad de calentar el “tip” o “needle” metálico y la cámara de ionización y/o la solución de analito que se introduce en flujo a través del “tip” (“needle”) (Fig 5).
|
|
|
|
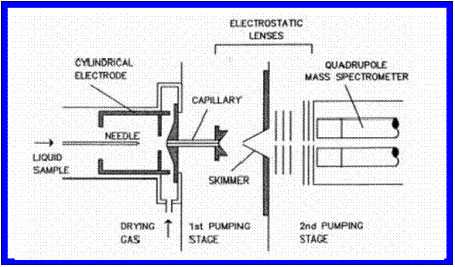

Por un lado eliminó el calentamiento que induce la descomposición de moléculas termolábiles. En segundo lugar modificó la forma de colectar y analizar a los iones producidos. En reemplazo de la caja de Faraday conectada al galvanómetro de Dole [3, 5-7], luego de colectar y obligar a pasar el haz de iones por un “primer agujero colector” (nozzle), y en segundo lugar “a través de una espumadera o separador de partículas sólidas” (skimmer) que terminaba de separar las moléculas gaseosas de aquéllas que pasan al estado sólido, introdujo a la corriente de moléculas gaseosas policargadas en un “quadruopolo” (Fig. 5). En esa época el “quadrupolo” ya se usaba exitosamente como un analizador de iones de muy buena resolución para rangos de m/z < 2000 Da. Se conocían muy bien su usos como filtro de iones y como elemento focalizador de haces de iones (Figs. 5 y 6) [2,3].
|
|
En forma casi simultánea, el grupo liderado por el profesor Aleksandrov en Rusia efectuó una mejora muy similar al ya conocido método de Dole, con el mismo objeto de mejorar la generación de moléculas gaseosas iónicas a partir de soluciones de analitos termolábiles [8].
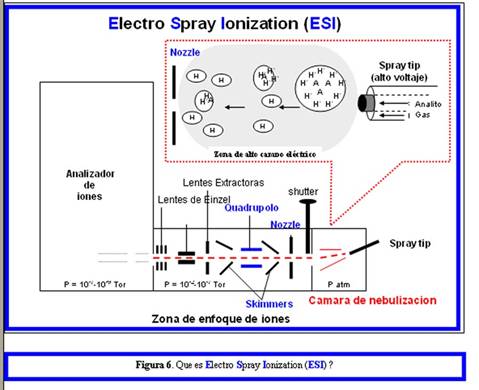
Como se muestra en la Fig. 6, la cámara de ionización ESI consta siempre de dos partes: (i) la cámara de nebulización y (ii) la zona de enfoque de iones. La primera se encuentra a presión atmosférica la segunda con vacío intermedio (10-2-10-4 Tor). En la primera al “tip” se le aplica alto voltaje (2-4 kV), en la segunda siempre están presentes “nozzle”, “skimmers” y “quadrupolo”. Según sea el signo del voltaje aplicado en el “tip” y en el “nozzle’ se analizan cationes (modo positivo) o se analizan aniones (modo negativo).
Acerca de proceso ESI
El proceso ESI está gobernado por una serie de parámetros química y físicos cuya optimización en forma conjunta determina la eficiencia del proceso. El proceso en su conjunto, del principio al final, puede representarse como un circuito eléctrico que genera y controla la nube (spray) de gotitas líquidas cargadas (Fig. 1). El proceso se inicia con el analito generalmente cargado (protonado) y disuelto en un medio con alto contenido acuoso (solvente polar). Al final del proceso el mismo analito como una molécula gaseosa multicargada y desnuda (desolvatada, no interactuado con el solvente) es atraída y colectada por el orificio de entrada del analizador de masas (nozzle) (Figs 1, 5 y 6). En el alto vacío del analizador (Fig. 6), el analito gaseoso policargado será selectivamente caracterizado por su relación m/z, quedando totalmente en el olvido el hecho de que se trate de un analito de alto peso molecular y termolábil.
Siguiendo con el proceso representado en las Figs. 1 y 3, las pequeñas gotitas inicialmente formadas tienen carga positiva o negativa, dependiendo de la polaridad asignada a la punta metálica del conducto inyector de la solución (electrodo madre del circuito; “tip”). El conjunto de moléculas protonadas o unidas a cationes metálicos (cationes) o deprotonadas (aniones) gaseosas quedan totalmente libres de solvente luego que éste se evapora. Como se muestra en la Fig. 4 en el caso de formarse especies protonadas, el número de cargas es igual al número de protones fijados a los grupos accesibles protonables. Según sea la naturaleza química y el peso molecular del analito, el número z puede variar entre 2 y 40 o ser aún mayor. Si el analito es una especie química única, el espectro resultante mostrará una serie regular de señales que reflejan los diferentes valores de m/z (Fig. 1). El hecho de que se generen familias de señales agrega complejidad a la interpretación de los espectros de masa obtenidos e inicialmente fue un factor de confusión que generaba rechazo en los posibles usuarios.
Había que pasar desde la imagen clásica de los espetros de masa obtenidos mediante otras técnicas de ionización, donde predominan los iones moleculares monocargados a las nuevas imágenes donde cada ión molecular intacto puede generar una familia de iones moleculares intactos policargados.
Sin embargo, se debe a Fenn no solamente el desarrollo tecnológico que optimizó el proceso “free-jet expansion” (y como consecuencia la eficiencia de la volatilización / ionización sin necesidad de calentar el sistema), sino que, además, fue él quién desarrolló los primeros programas de cálculo. Los que mostraron que la complejidad debida a las múltiples señales obtenidas por cada ión molecular, agregaba información para un cálculo más exacto del peso molecular del ión molecular intacto monocargado no detectado (valor de m). Este “secreto” fue revelado por Fenn como la “teoría de la carga múltiple” en una publicación del año 1987 [9]. Esta teoría demostró que los diferentes estados cargados del mismo ión molecular podían intrepretarse como medidas independientes del peso molecular del mismo analito y que, por lo tanto, un método de promediación basado en la solución simultánea de una familia de ecuaciones podría proveer una estimación mucho más precisa del peso molecular de macromoléculas.
Como se ve en la Fig. 1, la proteína de peso molecular 47.342 Da es analizada por ESI-MS. El espectro de masa resultante mostró 50 picos debidos a 50 valores diferentes de z (50 cargas diferentes). Las señales resultantes aparecen en la región de m/z 1-2000 Da y pueden ser analizadas muy fácilmente en cualquier analizador de masas. Este conjunto de datos por resolución del sistema de ecuaciones lleva a calcular un peso molecular muchísimo más exacto que el que se obtendría a partir de un solo ión.
Cabe señalar además, que otra de las ventajas del método de ionización ESI es que es el método de ionización de uso actual en espectrometría de masa menos invasivo y modificante de las propiedades del analito, salvo en lo que refiere a su volatilización y ionización. Por dicha razón es posible estudiar complejos moleculares debidos a débiles interacciones no-covalentes nativas tales como, por ejemplo: complejos proteína-proteína, enzima-substrato o proteína-ligando (Tablas 1 - 2).
Con la técnica ESI no solamente queda salvado el problema de la termolabilidad sino que además queda tecnológicamente resuelto el problema de volatilizar / ionizar analitos manipulando directamente la solución líquida que los contiene.
El uso de la espectrometría de masa como detector de alta sensibilidad y resolución de técnicas cromatográficas en fase líquida (LC) pasó a ser una realidad (Tabla 1).
ESI vs nano-ESI vs Z-ESI
Ya el mismo Fenn, en sus primeros trabajos, mostró también que la sensibilidad del proceso no se incrementaba aumentando la concentración del analito en la muestra en flujo introducida al sistema. Por el contrario, si el flujo variaba su velocidad de microlito/min a nanolitro/min la sensibilidad del experimento aumentaba en varios órdenes [10-11]. Se puede alcanzar sensibilidad a nivel del átomo-mol simplemente disponiendo de un “tip” de diámetro menor y disminuyendo el flujo de introducción de muestra y ésta es la diferencia entre la técnica denominada ESI y nano-ESI; la primera trabaja con un flujo de microlitro/min y la segunda de nanolitro/min. Además, cuando se trabaja con el “tip” nano-ESI, éste se lo acerca más al “nozzle” y como consecuencia toda la nube entra a la región de focalización, aumentándose la sensibilidad del método. Mayor sensibilidad y requerimientos 1000 veces menores de muestra !!! (Fig. 7).
|
|

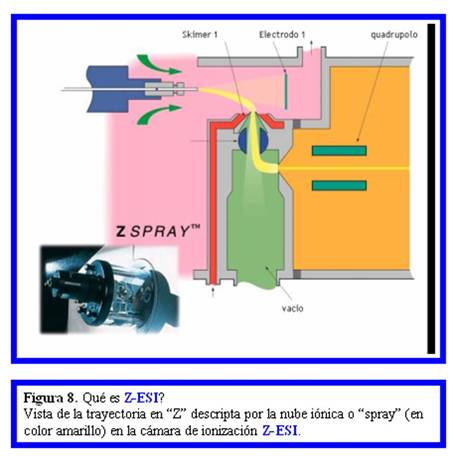
Una modificación tecnológica posterior, denominda Z-ESI,
tiene por objeto disminuir el número de partículas sólidas que llegan al
“quadrupolo” ubicado en la región de enfoque de iones (Fig. 6). Estas partículas se forman durante
la nebulización siendo una de las causas la presencia de sales en la solución. Como se muestra
en la Fig. 8 mediante un arreglo ortogonal del primer electrodo
orientador (electrodo 1) y el primer “skimmer” se desvía una parte de la nube
iónica inicial y se la redirecciona mediante un segundo “skimmer” de manera de
introducirla al “quadrupolo” de enfoque. En la actualidad existen tanto cámaras
de ionización Z-ESI (sinónimo de trabajo a escala del microlitro/min) como
cámaras de ioniación nano-Z-ESI.
|
|
Algunos detalles sobre equipamiento con ESI-MS disponible y aplicaciones...
Lo que diferencia drásticamente a los dos métodos de ionización suaves útiles para la volatilización / ionización de moléculas termolábiles, UV-MALDI y ESI, y a su vez al segundo de todos los demás métodos de ionización conocidos (EI, CI, FAB, FI, FD, etc. Ver detalle en [1]), es que en éste se generan iones moleculares gaseosos policargados y que el número de cargas (z) es casi proporcional al número de grupos polares presentes y al peso molecular. Los iones gaseosos policargados resultantes, normalmente, son iones moleculares intactos de masa m, cuya relación m/z está dentro de los valores que los analizadores convencionales usados en MS hasta fines del 80 podían manipular (colimar, enfocar, frenar, acelerar y detectar) eficientemente [2,12-13]. Por ejemplo, el análisis UV-MALDI-MS de la proteína comercial albumina bovina (pm 66266 Da) muestra una única señal a m/z 67266 (Da) [(M+H)+], mientras que el análisis por ESI-MS muestra una familia de 25 señales comprendidas entre m/z 1658 y 1020, con 40 < z < 65 [14].
Por la razón antes señalada, analizadores de uso rutinario en MS de moléculas termoestables de bajo peso molecular, en general, moléculas orgánicas, fueron exitosamente acoplados al ESI. Entre ellos basta mencionar el “quadrupolo” (Q), la combinación de sectores eléctricos y magnéticos (E-B), el “quadrupolo tridimentional” o “trampa iónica” (IT) y la “trampa iónica” combinada con un alto campo magnético denominado “fourier transform-ion ciclotron resonance” (FT-ICR) [1,2,12]. Este hecho determinó un rapidísimo desarrollo de la técnica ESI-MS, a tal punto que su rápida y eficaz utilización en el análisis de biomoléculas, hizo pensar a principios y mediados de la década del 90 que era de muchísima mayor utilidad en este campo que el UV-MALDI-MS.
El uso combinado de los analizadores, antes mencionados, para estudiar los procesos de fragmentación de moléculas pequeñas y la secuenciación de estos procesos ha sido muy desarrollado en las espectrometrías de masa convencionales (espectrometría de masa en tándem, MS/MS y en general MSn). El ensamble del ESI con dichos analizadores en tándem hizo surgir rápida y fácilmente la posibilidad de estudiar (i) la determinación de peso molecular de biomolécula intacta, (ii) la fragmentación del ión molecular de la biomolécula y (iii) la fragmentación selectiva de los iones primarios formados. De esta manera se pudo usar desde los inicios la ESI-MS tanto para determinar peso molecular (experimentos donde se genera el ión molecular intacto) como para determinar estructura química (experimentos donde se induce la fragmentacion del ión molecular) (uso de CAD; CID; “electron capture dissociation” ECD, “infra-red multi photon decomposition” IRMPD; ver modos de inducir fragmentación en la primer parte de este artículo [1]). En el análisis de proteínas y polipéptidos, el estudio de la fragmentación de las distintas generaciones de iones formados (fragmentación de ión molecular o ión padre, fragmentación del hijo, fragmentación del nieto, etc., etc.) permite determinar la estructura primaria o secuencia de los aminoácidos (Tabla 2) [1, 15-16]. En realidad la exitosa irrupción de la espectrometría de masa en el campo de la proteómica se inició por medio de la ESI-MS, siguiendo el protocolo denominado “bottom-up” (Tabla 2) [1, 15-16]. Cabe señalar que en la actualidad en el campo de la proteómica, además del protocolo “bottom-up” se usa el protocolo “top-down” efectuándose el análisis sobre la proteína intacta y su fragmentación sucesiva total dentro del espectrómetro de masa. Los estudios “top-down” se realizan sólo en equipos con ionización ESI del tipo ESI/FT-ICR con excitación por ECD y IRMPD y son clave para el estudio de modificaciones post-transduccionales (ver Tabla 2).
Tabla 2. Comparación del actual uso de ESI-MS y UV-MALDI-MS en el análisis de proteínas.
|
|
Proteína degradada enzimáticamente |
Proteína intacta y Modificaciones post-transduccionales |
Complejo proteico |
||||
|
|
del analito |
|
2D-LC/MS (ifenado) |
“Bottom-up” (“Genoma” y “A novo”) |
|
“Top-down”
a
|
|
|
-Mezclas de proteínas. |
(sobre la mezcla) |
|
|
|
|
|
|
|
(luego de LC ó 2D-GE) |
|
|
|
|
|
a Ver detalles en ref. [19].
b Ver detalles en ref. [19].
Equipos del tipo ESI-triple “quadrupolo” (ESI-QqQ o ESI-QQQ o ESI-tripleQ) fueron de los primeros que permitiron hacer proteómica, con el protocolo “bottom-up”.
La sensibilidad, resolución y exactitud de los ESI-MS varía según las marcas y los modelos. En la actualidad existen equipos más sofisticados donde al método de ionización ESI se lo combina con analizadores también únicos que permiten hacer los dos experimentos antes mencionados (sin fragmentación y con fragmentación). Estos analizadores son la denominada “trampa iónica” (IT) y el llamado “fourier transfor-ion ciclotron resonance” (FT-ICR) y los equipos serian ESI-IT y ESI-FT-ICR [2,12,16]. La resolución, exactitud y sensibiliad son muy altas. También existen los equipos que tienen en un arreglo en “tandem” dos analizadores de iones diferentes y a los experimentos realizados se los denomina “espectrometría de masa tandem” (MS/MS o MS2 ). El equipo clásico es el denominado ESI-Q-TOF que combina un “quadrupolo” (Q) y el analizador TOF de alta sensibilidad y también existe la combinación ESI-Q-IT [2].
Existen también equipos que combinan el “analizador de tiempo de vuelo” del tipo ESI-TOF, donde el espectrómetro de masa tiene solamente como analizador un “tiempo-de-vuelo” (TOF) y los experimentos se realizan directamente en modo “reflectrón” (trayectoria de iones: V; mayor exactitud en m/z, [1]), siendo útiles estos equipos sólo para determinar el peso molecular (condiciones experimentales de no fragmentación), ya que no tienen la opción de trabajar en el modo de operación que se denomina PSD (“post source decomposition”, ver detalles en [1]). También los hay con la combinación Q-TOF con un analizador TOF de mayor resolución, de doble reflejo (trayectoria de iones: N) y de triple reflejo (trayectoriade iones:W). En estos casos es posible efectuar estudios de secuenciación ya que el “quadrupolo” (Q) filtra o selecciona al ion padre cuya fragmentación se va a analizar dentro de TOF.
Todas las combinaciones intrumentales anteriores admiten el uso tanto de ESI como de nano-ESI y, en ambas escalas de trabajo, de la variante Z-ESI (Figs. 7-8).
Todos estos equipos tienen aún en la actualidad la seria limitación de que el rango dinámico de operación está limitado por el “quadrupolo” que como elemento de enfoque de iones forma parte de la unidad ESI (Fig. 6). Teóricamente este rango puede llegar a los 10000 Da. Sin embargo, en la mayoría de los equipos comerciales este rango tiene una cota superior de 4000 a 6000 Da (Tabla 1). Tener presente esta limitación instrumental es clave para entender que en un experimento ESI-MS, independientemente del analizador de iones utilizado, la no obtención de señales simplemente puede deberse a que los iones formados tienen valores de m/z que caen fuera del rango dinámico del instrumento.
Cálculo de m y z .¿Quién los efectúa? ¿Cuándo se efectúan?
Como ya se mencionó, el método de ionización ESI difiere de todos los demás [1] porque para un analito de peso molecular m, en lugar de detectarse una única señal a m/z con z = 1, se detecta una familia de señales con diferentes m/z siendo z > 1. Como se muestra en la Fig.9 en el ESI-MS de la mioglobina, analizado en modo positivo, no se detecta el ión molecular monocargado a m/z = 16951 Da sino que se detecta una familia de señales con valores de m/z comprendidos entre 808,221 y 1413,631 Da. Estos son iones policargados por poliprotonación [M(H)n]n+ , con valores de z comprendidos entre 21 y 12 (m/21 = 808,221 y m/12 = 1413,631 Da).
|
|
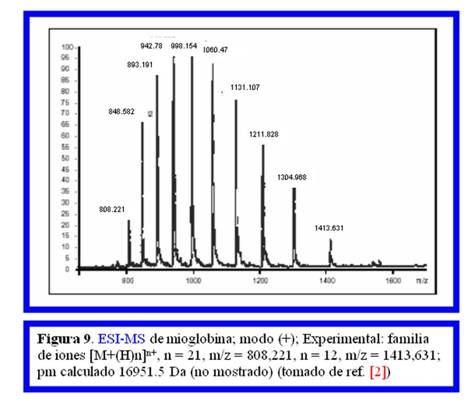
O sea que en realidad se obtiene una serie del tipo [M+nH]n+ , [M+(n+1)H](n+1)+ , [M+(n+m)H](n+m)+
Como se ve, se genera una serie de señales debidos a “quasi-iones moleculares” que difieren una de otra en un hidrógeno en su “quasi-peso molecular” m y en unidad de carga en su valor de z.
Si conocemos el valor de n, el problema está resuelto!
Afortunadamente estos cálculos se efectúan en forma automática durante el experimento, mediante programas de cálculo incluidos en el "soft" del equipo ESI-MS.
Básicamente los programas tienen en cuenta para un posible valor de M, y una dada combinación de átomos constituyentes de la molécula M, cúal es la relación teórica de intensidades para cada señal de los iones “cuasi-moleculares” teniendo en cuenta la abundancia isotópica natural de los elementos componentes. Para ese valor de M y esa combinación de átomo constituyentes trata de encontrar el valor de n que mejor calcula la distancia entre dos señales consecutivas.
Aquí aparece claramente cuan importante es conocer el grado de pureza del analito y de la solución para saber qué especie es la responsable de la carga de los iones “cuasi-moleculares”. El sistema de ecuaciones es diferente si la series incluye sólo al H+ ( [M+nH]n+ , [M+(n+1)H](n+1)+ ……… [M+(n+m)H](n+m)+), o si incluye sólo al Na+ ( [M+nNa]n+ , [M+(n+1)Na](n+1)+ ……… [M+(n+m)Na](n+m)+), o si incluye a ambos y más complicado es aún el sistema de ecuaciones si hay otros cationes en el medio. Esto es la denominada “baja tolerancia de sales” o “interferencia de sales” en la espectrometría de masa ESI (Tabla 1).
El programa efectúa en forma iterativa estos cálculos tantas veces como sea necesario hasta encontrar el valor óptimo de n. O sea aquél que daría por simulación un espectro ESI idéntico al obtenido experimentalmente. Y…de esta manera, “mágicamente” aparecen en pantalla los valores de z al lado de cada señal del espectro mientras se está efectuando el experimento.
Sólo hay que darle al programa como información (i) el umbral de sensibilidad del experimento, (ii) quién es la especie responsable de la carga (H+ y/o catión metálico) y (ii) cuáles serían los elementos constituyentes del analito, de manera de evitar que el programa “por default” considere todas las combinaciones posibles de la Tabla Periódica!
De esta manera se obtiene no solamente el valor de n sino además el valor del peso molecular del ión molecular intacto, M.
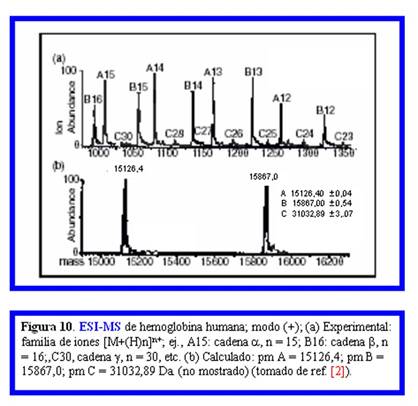
Como se muestra en la Fig. 10 para la hemoglobina humana, el análisis por ESI-MS en modo positivo mostró la familia de iones policargados para cada una de las cadenas (A, alfa; B, beta; G, gama) indicando al lado de cada señal el valor de z calculado ((a) espectro obtenido experimentalmente). En la parte (b) se muestra la ubicación de los iones moleculares monocargados correspondientes a las cadenas A y B en el espectro simulado por el mismo programa de cálculo.
|
|
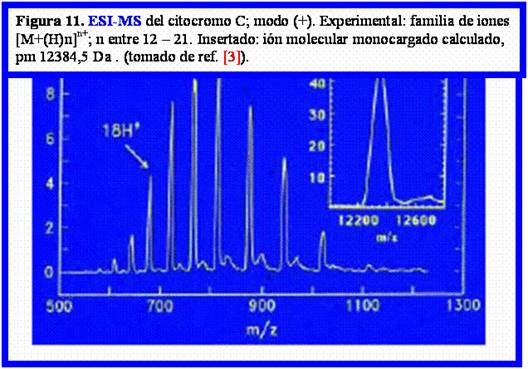
En la Fig. 11 se muestra el primer ejemplo descripto por Fenn [3] de cálculo del peso del ión molecular monocargado de una proteína (citocromo C, pm 12384,5 Da, espectro insertado) cuyo espectro de masa ESI mostró, en modo positivo, la familia de iones policargados debido a poliprotonación, con m/z en el rango 600 – 1100 Da (z entre 12 – 21).
|
|
Para terminar ...complejos proteicos y polímeros sintéticos
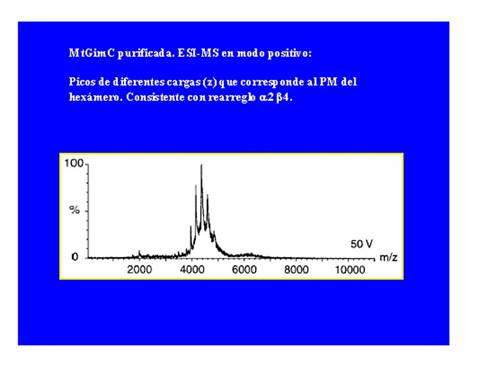
Finalmente, para complementar en imágenes el panorama de aplicaciones actuales de la técnica ESI-MS [3,12-13,15-16], en las Figs 12-13 se muestran complejos proteicos en la primera [17] y polímeros sintéticos del tipo silsesquioxanos en la segunda [18]. En la Fig. 12 (imagen superior) se muestra, el espectro obtenido en modo positivo para el complejo proteíco nativo de la chaperona MtGimC aislada y purificada y en la Fig. 12 (imagen inferior) los espectros obtenidos analizando sucesivamente alícuotas de una solución que ha sido preparada mezclando cantidades equimoleculares de las unidades proteicas alfa y beta que es sabido que forman el complejo proteico nativo [17]. Esta interesante aplicación se basa en la ya mencionada particularidad de la ESI que es la de mantener entre las unidades proteicas gaseosas policargadas las mismas interacciones intermoleculares que están presentes en la solución. Por supuesto que esto es válido no sólo para proteínas, sino para analitos en general.
|
|
|
|

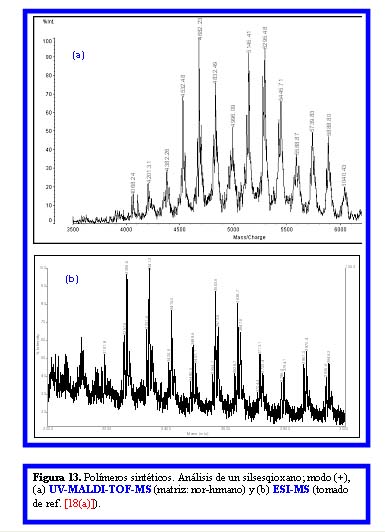
En cuanto a sus aplicaciones a polímeros sintéticos, en la Fig 13 se muestran los espectros de masa UV-MALDI-TOF y ESI obtenidos en modo positivo para un polímero sintético. Este contiene en su estructura Si (silsesquioxanos) y del espectro UV-MALDI (Fig. 13(a)), se concluye que su peso molecular promedio es del orden de 5000 Da, coincidiendo este valor con el obtenido por SEC (cromatografía de exclusión por tamaño, [18a]). En el mismo espectro se observa la distribución gaussiana esperada de los oligómeros constituyentes (información cuantitativa). Por el contrario, el espectro ESI (Fig. 13(b)) muestra a primera vista un aspecto totalmente diferente. No solamente las señales aparecen a valores de m/z menores debido a la policarga (ESI, m/z entre 2200 – 3000 Da; UV-MALDI, m/z entre 4000 –6500 Da) sino que, además, se pierde la imagen de distribución gaussiana de los oligómeros debido al entrecruzamiento de señales de los diferentes oligómeros policargados. Con este último ejemplo se muestra claramente que en el análisis de mezclas en general, sean o no éstas oligómeros de polímeros naturales o sintéticos, se requiere para la interpretación de la información obtenida por ESI-MS disponer de los espectros obtenidos por UV-MALDI-MS.
|
|
Referencias
[1] Erra-Balsells, R., abril 2004. Del vuelo de las proteínas y de cómo
lograrlo (espectrometría de masa UV-MALDI). Quimica Viva, 3(2).
[2] Back to Basics Manual,
Micromass UK Limited, www.micromass.co.uk
[3] Fenn, J. B., Electrospray
Wings for Molecular Elephants, Nobel Lecture, http//www.nobel.se
[4] Bothner, B., Dong, X. F.,
Bibbs, L., Johnson, J. E. and Siuzdak, G., 1998. Evidence of viral capsid
dynamics using limited proteolysis and mass spectrometry. J. Biol. Chem. 273,
673-676.
[5] Dole, M., Mach, L. L.,
Hines, R. L., Mobley, R. C., Ferguson, L. D. and Alice, M. B., 1968. Molecular
beams of macroions. J. Chem. Phys. 49, 2240-2247.
[6] Mann, M., Meng, K. C. and
Fenn, J. B., 1988. Proc. Proceedings of the 36th Annual Conference on Mass
Spectrometry and Allied Topics, Am. Soc. For Mass Spectrom.,
[7] Yamashita, M. and Fenn, J.
B., 1984. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 88,
4451-4469.
[8] Aleksandrov, M. L., Gall, L.
N., Krasnov, V. N., Nikolaev, V. I., Pavlenko, V. A. and Shkurov, V. A. 1984. Dokl Akad Nauk
SSSR. 277, 379-383.
[9] Fenn, J. B., Mann, M.,
Meng, C. K., Wong, S. F. and Whitehouse, C. M., 1989. Electrospray ionisation
for mass spectrometry of large biomolecules. Sciences 246, 64.
[10] Wilm, M., Shevchenko, A.,
Houtaeve, T., Breit, S., Schweigerer, L., Fotsis, T and Mann, M. 1996.
Femtomole sequencing of porteins from polyacrylamide gels by nano-electrospray
mass spectrometry. Nature 379, 466-469.
[11] Valaskovic, G.A. and
McLafferty, F. W. 1995. Attomole-sensitivity electrospray source for
laarge-molecule mass spectrometry. Anal. Chem. 67, 3802-3805.
[12] Cole, R. B. 1997.
Electrospray Ionization Mass Spectrometry, Fundamentals, Instrumentation, and
Applications, John Wiley and Sons, INC., NY.
[13] Fenn, J. B., Mann, M.,
Meng, C. K., Wong, S. F. and Whitehouse, C. M., 1990. Electrospray
ionization-principles and practice. Mass Spectrom. Rev. 9, 37-70.
[14] Smith, R. D., Loo, J. A.,
Edmonds, C. G., Barinaga, C. J. and Udseth, H. R., 1990. New developments in
biochemical mass spectrometry: electrospray ionization. Anal. Chem., 62,
882-889.
[15] (a) Liebler, D. C. 2002,
Introduction to Proteomics- Tools for the New Biology, Human Press, Totowa, NJ; (b) Mann, M. and Jensen, O.
N., 2003. Proteomic analysis of post-translational modifications
(www.nature.com/naturebiotechnology) Vol 21.
[16] Siuzdak, G., 1996. Mass
Spectrometry for Biotechnology, Academic Press.
[17] Fanderich, M., Tito, M.
A., Leroux, M. R., Rostom, A. A., Ulrich Hartl, F., Dobson, C. M. and Robinson,
C. V., 2000. Observation of the noncovalent assembly and disassembly pathways
of the chaperone complex MtGimC by mass spectrometry, Proc. Natl. Acad. Sci.
USA 97, 14151-14155.
[18] (a) Fasce, D. P.,
Williams, R. J. J., Erra-Balsells, R., Ishikawa, Y. and Nonami, H., 2001.
One-Step Synthesis of Polyhedral Silsesquioxanes Bearing Bulky Substituents:
UV-MALDI-TOF and ESI-TOF Mass Spectrometry Characterization of Reaction
Products. Macromolecules 34, 3534-3539; (b) Williams, R. J. J., Erra-Balsells,
R., Ishikawa, Y., Nonami, H., Mauri, A. N. and Riccardi, C. C., 2001.
UV-MALDI-TOF and ESI-TOF mass spectrometry characterization of silesquioxanes
obtained by the hydrolytic condensation of (3-glycidoxypropyl)trimethoxysilane.
Macromol. Chem. Phys. 202, 2425-2433; (c) Montaudo, M. S., 2002. Mass Spectra
of Copolymers. Mass Spectrom. Rev. 21, 108-144.
[19] (a) Kelleher, N L., Lin,
H. Y., Valaskovic, G. A., Aaserud, D. J., Fridriksson, E. K., McLafferty, F.
W., 1999. Top down versus Bottom up Protein Characterization by Tandem
High-Resolution Mass Spectrometry. J. Am. Chem. Soc. 121, 806-812; (b) Oh, H.
B., Breuker, K., Sze, S. K., Ge, Y., Carpenter, B. K. and McLafferty, F. W.,
2002. Secondary and Tertiary Structures of Gaseous Protein Ions Characterized
by Electron Capture Dissociation Mass Spectrometry and Photofragment
Spectroscopy. Proc. Natl. Acad. Sci. USA 99, 15863-15868; (c) Breuker, K. and
McLafferty, F. W., 2003. Native Electron Capture Dissociation for Structural
Characterization of Noncovalent Interactions in Native Cytochrome c. Angew.
Chem. Int. Ed. 42, 4900-4904; (d) Patrie, S. M., Charlebois, J. P., Whipple,
D., Kelleher, N. L., Hendrickson, C. L., Quinn, J. P., Marshall, A. G.,
Mukhopadhyay, B., 2004. Construction of a Hybrid Quadrupole/Fourier transform
ion Cyclotron Resonance Mass Spectrometer for Versatile MS/MS Above 10 kDa. J.
Am. Soc. Mass Spectrom. 15, 1099-1108.
*Dra. Rosa Erra-Balsells. E-mail: erra@qo.fcen.uba.ar
|
|
Revista QuímicaViva Número 3, año 3, septiembre 2004 quimicaviva@qb.fcen.uba.ar |