Síndrome de Rett: una enfermedad del neurodesarrollo de diagnóstico esquivo
María Belén Cardillo, Bruno G. Berardino y Eduardo T. Cánepa
Laboratorio de Neuroepigenética y Adversidades Tempranas, Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires e IQUIBICEN, CONICET, Argentina.
Recibido: 01/08/2024 - Aceptado: 18/08/2024
Resumen
El síndrome de Rett es una severa enfermedad neurológica que afecta mayoritariamente a mujeres causada, en el 95% de los casos, por mutaciones de novo en el gen MECP2. La pérdida de función de la proteína MECP2 deriva en un deterioro progresivo en la salud física y mental de las niñas afectadas. Sobre la base de estudios funcionales, se ha propuesto que MECP2 regula la transcripción génica, la compactación de la cromatina y el procesamiento mRNA y RNA no codificantes. Si bien esta proteína cumple un rol importante en diversos tejidos es en el cerebro donde desempeña un papel fundamental que se refleja en la gravedad de las manifestaciones clínicas que tienen su origen en ese órgano.
Actualmente, el diagnóstico del síndrome de Rett continúa siendo clínico y se basa principalmente en la historia de regresión del desarrollo y muy frecuentemente es tardío y en general subdiagnosticado. Este retraso en el diagnóstico tiene consecuencias negativas graves y una de las más importantes es la pérdida de oportunidades para intervenciones tempranas. Con el objetivo de atenuar esta necesidad hemos diseñado y comenzado a desarrollar un proyecto que consiste en evaluar la expresión diferencial de microRNA en sangre de las niñas afectadas por el síndrome de Rett a fin de explorar su potencial como biomarcadores tempranos de la enfermedad y de la severidad de las manifestaciones clínicas. Además, analizar la expresión de microRNA en las vesículas extracelulares derivadas de neuronas de modo de revelar procesos biológicos potencialmente alterados por la enfermedad e iluminar nuevos sitios de intervención terapéutica. Mejorar la calidad de vida de las niñas afectadas subyace a los objetivos experimentales.
Palabras clave: biomarcador diagnóstico y pronóstico; enfermedades del neurodesarrollo; cerebro; microRNA; vesículas extracelulares
Rett syndrome: a neurodevelopmental disease with an elusive diagnosis
Summary
Rett syndrome is a severe neurological disorder that primarily affects females and is caused, in 95% of cases, by de novo mutations in the MECP2 gene. The loss of function of the MECP2 protein leads to a progressive deterioration in the physical and mental health of affected girls. Based on functional studies, it has been proposed that MECP2 regulates gene transcription, chromatin compaction, and the processing of mRNA and non-coding RNA. While this protein plays an important role in various tissues, it is in the brain where it performs a fundamental role, as reflected in the severity of clinical manifestations originating in this organ. Currently, the diagnosis of Rett syndrome remains clinical, primarily based on the history of developmental regression, and it is often late and generally underdiagnosed. This delay in diagnosis has serious negative consequences, one of the most important being the loss of opportunities for early interventions. To address this need, we have designed and begun to develop a project that involves evaluating the differential expression of microRNA in the blood of girls affected by Rett syndrome to explore their potential as early biomarkers of the disease and the severity of clinical manifestations. Additionally, analyzing the expression of microRNA in extracellular vesicles derived from neurons aims to reveal biological processes potentially altered by the disease and highlight new sites for therapeutic intervention. Improving the quality of life of affected girls underlies the experimental objectives.
Keywords: diagnostic and prognostic biomarkers; neurodevelopment diseases; brain; microRNA; extracellular vesicles
La enfermedad: sus causas y consecuencias
El síndrome de Rett es un severo y progresivo desorden neurológico que afecta el desarrollo y la función del cerebro principalmente en mujeres. Con una incidencia de 1:10 000–15 000, el síndrome de Rett es la segunda causa más común de discapacidad intelectual grave en mujeres.
En el 90-95% de los pacientes diagnosticados con Rett clásico, la enfermedad es causada por mutaciones de pérdida de función en el gen MECP2, ligado al cromosoma X, que codifica la proteína de unión a las secuencias del DNA CpG metiladas, MECP2. Estas son mutaciones de novo mayoritariamente con origen en las células germinales paternas [1, 2]. Por esta razón, el síndrome de Rett pese a ser una enfermedad genética no es hereditaria. Este origen paterno puede explicarse por una combinación de los elevados niveles de metilación y divisiones mitóticas en la línea germinal masculina. Las mutaciones en MECP2 en el único gen X de los varones generalmente conducen a encefalopatías congénitas severas y fallecen antes de los dos años [3].
Mutaciones en otros genes como CDKL5 y FOXG1 han sido asociadas con formas atípicas del síndrome de Rett [4, 5]. Sin embargo, aunque comparten algunos síntomas, el criterio médico tiende a considerarlos y tratarlos como enfermedades diferentes.
Características clínicas y genéticas
Cuatro manifestaciones clínicas constituyen las características diagnósticas principales del síndrome de Rett: pérdida del lenguaje expresivo; pérdida de las habilidades motoras finas (es decir, de las manos); deterioro de la deambulación; y presencia de movimientos estereotípicos de las manos. Estos rasgos característicos, y otros más variables en frecuencia y gravedad, surgen en diferentes momentos durante el curso dinámico del síndrome de Rett [1]. El síndrome de Rett es la segunda causa más común de discapacidad intelectual grave después del síndrome de Down.
La enfermedad se desarrolla en cuatro etapas. Después de un período posnatal temprano relativamente neurotípico, los primeros síntomas sobrevienen entre los 6 y los 18 meses de vida, seguido de una segunda etapa de pocos meses durante la cual la desaceleración del crecimiento de la cabeza y el retraso cognitivo y motor global se vuelven evidentes junto con una pérdida variable del lenguaje hablado, del contacto visual y de las habilidades manuales. La regresión del desarrollo es la característica diagnóstica distintiva del síndrome de Rett [6]. Hay una tercera etapa de meseta en la evolución de la enfermedad de duración variable, seguida luego de un fuerte deterioro y la pérdida de la capacidad ambulatoria.
Desde un punto de vista estructural y funcional, MECP2 es un ejemplo de una proteína intrínsecamente desordenada dado su contenido relativamente bajo de organización estructural secundaria y terciaria en solución. Pese a esto, la proteína se puede dividir en varios dominios estructurales/funcionales bien definidos: un dominio N-terminal (NTD), un dominio de unión a metilos (MBD), dominio de represión transcripcional (TRD), región interviniente (ID), un dominio de interacción con el correpresor NCoR (NID) y un dominio C-terminal (CTD). El TRD incluye la secuencia de localización nuclear (NLS) [7].
El síndrome de Rett se asocia con más de 500 mutaciones patógenas o probablemente patógenas de MECP2 que afectan principalmente a los exones 3 y 4 y a la región C-terminal [8]. Sin embargo, ocho mutaciones puntuales de MECP2, cuatro de ellas con pérdida de sentido y cuatro sin sentido (R106W, R133C, T158M, R168X, R255X, R270X, R294X y R306C); deleciones C-terminales; y deleciones exónicas representan la gran mayoría de los casos [9, 10]. El mapeo de mutaciones en la estructura cristalina de la proteína revela que la mayoría de estas mutaciones se encuentran en tres dominios clave: el dominio de unión a metilo (MBD), el dominio de represión transcripcional (TRD) y el dominio de señal de localización nuclear (NLS) [8].
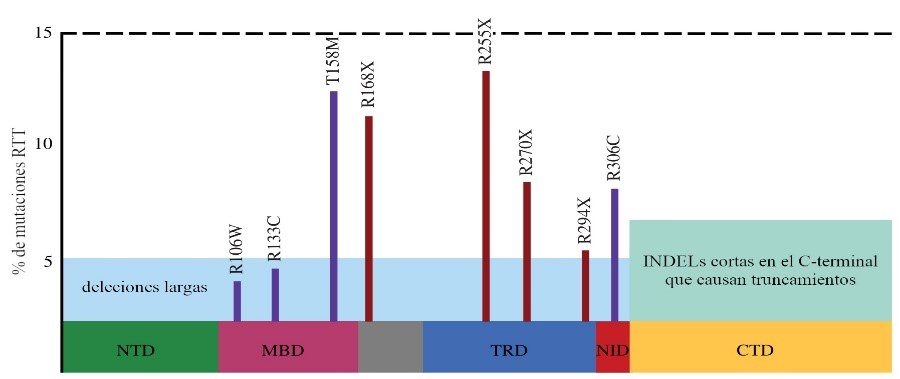
Figura 1: Mutaciones más frecuentes en la proteína MECP2 reportadas en pacientes con síndrome de Rett. .La figura indica la ubicación de las mutaciones en MECP2 respecto a los diferentes dominios de la proteína y la frecuencia con que se encuentra cada una de ellas en pacientes afectados con el síndrome de Rett (adaptado de [11]). Las abreviaturas se encuentran explicadas en el texto.
No hay una correlación estrecha entre el genotipo y el fenotipo para la enfermedad, aunque se reconocen algunos perfiles generales. El grupo que presenta sintomatología más severa incluye en general mutaciones sin sentido que afectan el dominio de represión transcripcional [12, 13].
Efectos moleculares de MECP2:
interacción con el DNA y el RNA
Si bien MECP2 se expresa de forma ubicua se encuentra muy enriquecida en el cerebro y la pérdida de su función afecta a diferentes tipos de células en múltiples regiones cerebrales. La deleción específica de MECP2 en el cerebro de ratones reproduce fenotipos clave del síndrome de Rett, como movimiento reducido, marcha anormal, agarre de las extremidades traseras, anomalías respiratorias y letalidad temprana, los cuales también están presentes en ratones KO para MECP2 [14]. Estos hallazgos destacan el papel crucial de MECP2 para la funcionalidad del cerebro.
En el contexto de la cromatina, las marcas epigenéticas sobre el DNA y las histonas, el principal componente proteico de la cromatina, son agregadas o eliminadas por una gran cantidad de proteínas con variadas actividades enzimáticas denominadas genéricamente escritoras o borradoras, respectivamente [15, 16]. Otras proteínas, cofactores que regulan la transcripción, interaccionan con estas marcas epigenéticas y son denominadas lectoras [17]. Este último es el caso de MECP2, una proteína lectora de la metilación del DNA. La interacción con el DNA es una parte esencial de la función de MECP2, ya que es principalmente una proteína nuclear de unión a la cromatina que actúa como una lectora epigenética. Sobre la base de estudios funcionales, se ha propuesto que MECP2 regula la transcripción génica, la compactación de la cromatina y el procesamiento y expresión de mRNAs y RNAs no codificantes [18].
MECP2 tiene efectos espaciotemporales diversos según las regiones cerebrales, las fases de desarrollo y los tipos de células. MECP2 regula múltiples etapas del desarrollo cerebral, incluida la neurogénesis prenatal, el desarrollo posnatal de las conexiones y funciones sinápticas y el refinamiento de los circuitos neuronales en función de la experiencia. Al igual que muchos otros trastornos cerebrales, el síndrome de Rett se asocia con una disfunción que refleja un fallo funcional en varios niveles, incluidos los niveles de regulación genética, expresión molecular, función sináptica y circuitos neuronales, y en varias etapas del desarrollo.
Los mecanismos básicos por medio de los cuales MECP2 ejerce sus funciones se conocen desde hace tiempo, sin embargo, relacionarlos con los síntomas fenotípicos causados por las mutaciones encontradas en Rett es complicado debido los efectos pleiotrópicos de los genes que regula y la compleja trama de vías de señalización en las que está funcionalmente involucrada. MECP2 se identificó originalmente en función de su capacidad para unirse al DNA que contiene citosinas metiladas en los dinucleótidos CpG a través de su dominio de unión MBD lo que conduce a reprimir la expresión génica. Posteriormente, se determinó su capacidad para actuar también como un activador transcripcional según el contexto molecular [18, 19]. Más recientemente, se reportó que MECP2 interactúa también con secuencias CA metiladas promoviendo también la represión transcripcional. Funcionalmente, ahora está claro que la fosforilación dependiente de la actividad neuronal en residuos específicos de MECP2 media su interacción con varios correpresores transcripcionales. Entre ellos hay evidencias de que la interacción con el complejo correpresor HDAC-SIN3A o con HDAC3 y el receptor nuclear correpresor NCoR-SMRT son necesarios para mediar los efectos represivos de MECP2 [20, 21] compactando la estructura de la cromatina y removiendo los grupos acetilo de los residuos lisina de las histonas. En este sentido, mutaciones en MBD y/o NID de MECP2 que impiden el reclutamiento de los complejos correpresores, son relevantes en las manifestaciones clínicas de los pacientes afectados por el síndrome de Rett.
Como mencionamos, la fosforilación de MECP2 inducida por la actividad neuronal es importante para regular la interacción con los correpresores. La actividad neuronal induce la fosforilación de MECP2 en varios sitios, incluido T308 [22]. La fosforilación en T308 bloquea la interacción de MECP2 con el complejo NCoR–SMRT, suprimiendo así la capacidad de MECP2 para reprimir la transcripción. La mutación sin sentido asociada a Rett en MECP2, que conduce a una sustitución R306C, elimina la fosforilación de MECP2 en T308 derivando en la interrupción en la regulación de la interacción de MECP2 con el complejo NCoR en respuesta a la actividad neuronal.
Además de su capacidad de unirse al DNA metilado, estudios más recientes han revelado que MECP2 también interactúa con el RNA, agregando otra capa a sus funciones reguladoras, particularmente en el contexto del desarrollo y la función neuronal [23]. Estas interacciones con el RNA influyen en varios procesos postranscripcionales, lo que contribuye a la complejidad del papel de MECP2 en la regulación de la expresión génica. La proteína MECP2 tiene varios dominios que le permiten unirse directamente al RNA ubicados, principalmente, en las regiones ID y MBD. La actividad de unión del MECP2 al RNA parece ser relativamente promiscua, ya que se une a varias clases, incluidos los mRNA, los RNA largos no codificantes (lncRNA) y los microRNA (miRNA). Este amplio espectro de unión sugiere un papel versátil para el MECP2 en el metabolismo del RNA [24, 25]. También hay evidencias de que MECP2 puede regular el splicing alternativo de mRNA a través de su unión a los factores de splicing y al spliceosoma [26, 27]. Es importante destacar que la capacidad de MECP2 de interaccionar con estos factores reside principalmente en el extremo C-terminal, región de la proteína que se encuentra mutada en más del 10% de los casos de Rett sugiriendo que este mecanismo de procesamiento del RNA estaría afectado en estos pacientes.
Interacción entre MECP2 y miRNA
Entre los mecanismos epigenéticos los miRNA, una nutrida clase de moléculas con actividad regulatoria de la traducción y estabilidad de los mRNA, desempeñan un papel esencial en una variedad de procesos. Los miRNA son importantes en particular en el sistema nervioso interviniendo en la neurogénesis, la maduración del cerebro y la plasticidad sináptica [28].
Los niveles de expresión de miRNA se encuentran desregulados en los cerebros de los pacientes con síndrome de Rett y ofrecen una herramienta potencial para medir la progresión de la enfermedad y la respuesta al tratamiento [29]. De modo coincidente, se ha observado una desregulación en la expresión de miRNA en el cerebro en general y en regiones particulares como el hipocampo y la corteza en ratones nulos para MECP2, en cultivos de neuronas de ratones deficientes en MECP2 y en cultivos neuronales derivados de iPSC y organoides cerebrales generados a partir de individuos con síndrome de Rett [30].
La proteína MECP2 reprime la transcripción miRNA primarios (pri-miRNA) mediante su unión a sitios CG metilados en sus regiones promotoras. La deficiencia de MECP2 causa un aumento de la transcripción de pri-miRNA, lo que conduce a un aumento de los niveles de miRNA maduro. Por lo tanto, se espera que las mutaciones asociadas a Rett en la secuencia que codifica el MBD y TRD o NID de MECP2 conduzcan a aumentos en estos miRNA regulados por MECP2.
Además de su efecto sobre la transcripción de los miRNA, MECP2 puede inhibir su procesamiento. El procesamiento del pri-miRNA en el precursor (pre-miRNA) que es realizado por el complejo microprocesador nuclear antes de exportarlo al citoplasma es el paso regulador clave para determinar los niveles de miRNA maduro en la célula [31]. Las proteínas microprocesadoras centrales son DROSHA, que escinde el pri-miRNA y DGCR8 que proporciona afinidad de unión del RNA a DROSHA. La función de MECP2 consiste en unirse a la proteína microprocesadora DGCR8 a través del dominio C-terminal y, por lo tanto, obstaculizar la interacción de DGCR8 con DROSHA disminuyendo el procesamiento. En individuos afectados con Rett que portan mutaciones o truncamientos en el extremo C-terminal de MECP2, la unión entre MECP2 y DGCR8 está impedida, dejando a DROSHA libre para unirse y activar el procesamiento de los miRNAs conduciendo a un aumento desregulado de los niveles de miRNA en el citoplasma celular.
Estas evidencias acerca del papel que desempeña MECP2 en el procesamiento y expresión de los miRNA y otros RNA no codificantes sugieren que estas moléculas podrían constituir factores mediadores en las consecuencias epigenéticas derivadas de la pérdida de función de esta proteína, un importante modificador de la cromatina [32].
Tratamientos, terapias y ensayos clínicos
El tratamiento del síndrome de Rett se dirige a los síntomas específicos que se manifiestan en cada individuo. El tratamiento puede requerir los esfuerzos coordinados de un equipo de especialistas, pediatras, neurólogos pediátricos, gastroenterólogos, logopedas, psiquiatras, nutricionistas y otros profesionales de la salud para planificar sistemáticamente y de manera integral el tratamiento de un niño afectado. El plan de tratamiento específico debe ser personalizado. La intervención temprana del desarrollo es importante para garantizar que los niños afectados alcancen su potencial beneficiándose de la terapia ocupacional, física y del habla. El apoyo psicosocial para toda la familia también es esencial.
En 2023, la Administración de Alimentos y Medicamentos de los Estados Unidos aprobó la trofinetida (Daybue) como el primer tratamiento para el síndrome de Rett en adultos y niños de 2 años de edad o más, diseñada para aliviar los síntomas más severos de la enfermedad. En varias compañías farmacéuticas se están desarrollando estudios preclínicos y clínicos apoyados en diversas estrategias: reemplazo génico introduciendo variantes no mutadas del gen MECP2, edición génica por CRISPR, reactivación de MECP2 en el otro cromosoma X, el reemplazo de la sección de RNA donde se encuentran las mutaciones y la administración de la proteína salvaje al paciente.
Un problema y un proyecto
El problema: el diagnóstico tardío y sus consecuencias
Los síntomas principales de la afección del síndrome de Rett, luego de un período de desarrollo posnatal temprano típico, incluyen la detención del desarrollo cognitivo y motor, la pérdida de las habilidades verbales adquiridas y la aparición de movimientos repetitivos estereotipados de las manos [33]. Si bien estos síntomas comienzan a ser aparentes entre los 9 y 15 meses de edad, la enfermedad se diagnostica bastante más tarde. Según datos disponibles en EEUU, el síndrome de Rett se diagnostica a una edad promedio de 2,8 años [1]. En nuestro país las diferencias son más inquietantes. De acuerdo con lo manifestado por padres y madres de niñas afectadas con el síndrome de Rett, la edad promedio en la cual detectan los primeros síntomas de la existencia de problemas en el desarrollo de sus hijas es de 11 meses, mientras que la confirmación de que están afectadas por Rett, realizada mediante un análisis genético es, en promedio, a los 4,5 años. Como se ve, el lapso es muy grande y es tiempo perdido para la aplicación de tratamientos que atenúen los síntomas y las manifestaciones clínicas y de este modo aliviar y mejorar la calidad de vida de las personas afectadas por Rett.
Actualmente, el diagnóstico del síndrome de Rett continúa siendo clínico y se basa principalmente en la historia de regresión del desarrollo con las incertezas asociadas a los recuerdos de los padres o cuidadores; por lo que frecuentemente es tardío [1, 2]. Este retraso en el diagnóstico puede tener varias consecuencias negativas graves y de gran alcance. La presencia de mutaciones en MECP2 respalda, pero no confirma la severidad de la enfermedad, debido a las limitadas correlaciones genotipo-fenotipo en el síndrome de Rett.
Una de las consecuencias más importantes del diagnóstico tardío es la pérdida de oportunidades para intervenciones tempranas. Las investigaciones han demostrado que la estimulación sensoriomotora temprana e intensiva puede beneficiar en gran medida a los niños con síndrome de Rett, mejorando las funciones motoras y las capacidades cognitivas [33, 34]. Cuando el diagnóstico se retrasa, se pierden estas oportunidades críticas para la intervención, lo que conduce a peores resultados en las habilidades motoras, las capacidades de comunicación y la función cognitiva en general. De igual modo, una dieta alimenticia apropiada mejora la sintomatología de las niñas. Las familias de niños con síndrome de Rett suelen afrontar un proceso de diagnóstico prolongado y estresante. Este "peregrinaje" implica numerosas consultas médicas, diagnósticos erróneos y tratamientos innecesarios. El desgaste emocional y psicológico que sufren las familias puede ser inmenso y generar ansiedad, frustración y una sensación de impotencia. Un diagnóstico tardío prolonga este período de incertidumbre y angustia, lo que repercute en la salud mental y el bienestar tanto del paciente como de su familia.
Sin un diagnóstico adecuado, el manejo de los síntomas del síndrome de Rett puede ser complicado. Un diagnóstico tardío suele implicar que los síntomas no se atribuyan correctamente al síndrome de Rett, lo que lleva a tratamientos inadecuados o ineficaces. Por ejemplo, los movimientos repetitivos de las manos y las anomalías motoras pueden confundirse con otras afecciones, lo que da lugar a tratamientos que no abordan los problemas subyacentes. Las estrategias de manejo adecuadas, que incluyen fisioterapia personalizada, terapia ocupacional y medicamentos apropiados, solo se pueden implementar una vez que se realiza un diagnóstico definitivo.
Finalmente, el efecto acumulativo del diagnóstico tardío es una calidad de vida comprometida para el niño afectado. La incapacidad de comunicarse de manera efectiva, la movilidad reducida y la presencia de desafíos conductuales y cognitivos graves pueden conducir al aislamiento social y a una menor participación en las actividades cotidianas. El diagnóstico temprano permite una mejor planificación e implementación de medidas de apoyo que pueden mejorar la calidad de vida del niño, promoviendo una mayor independencia e integración social. Sumado a esto, la falta de un diagnóstico preciso del síndrome de Rett lleva a suponer muy fundadamente que hay muchas niñas que están afectadas por el síndrome y que no han sido diagnosticadas o que tienen un diagnóstico erróneo. De acuerdo con los datos oficiales provenientes del Registro de pacientes con enfermedades poco frecuentes (EPOF) hay alrededor de 400 personas con síndrome de Rett en nuestro país. Sin embargo, si observamos los números de España, un país con casi la misma población, hay registrados en la actualidad alrededor de 3000 casos.
Estas observaciones resaltan la necesidad de contar no solo con un diagnóstico temprano de la enfermedad sino también con fenotipos que permitan estimar la progresión de la enfermedad y que adquieran así un valor pronóstico de relevancia.
El proyecto
El comienzo con la Fundación SinRett
En nuestro país en 2021 fue creada la Fundación SinRett que reúne a los familiares de personas con síndrome de Rett con el objetivo de darle mayor visibilidad a la enfermedad y generar conciencia e inclusión para los pacientes. Aunque con sede en Rosario la fundación tiene alcance nacional y ha reunido hasta el momento más de 100 familias de todo el país. El proyecto de investigación que hemos comenzado en el Laboratorio de Neuroepigenética y Adversidades Tempranas se está llevando a cabo en colaboración con la Fundación y ha contado con el apoyo de ALAPA (Alianza Argentina de Pacientes) que ha otorgado una beca de investigación para un miembro del equipo de trabajo.
El contacto con Fabián Crespo, el presidente de la Fundación SinRett, nos permitió conocer la realidad de las niñas afectadas y de sus familiares quienes además de las consecuencias de la enfermedad deben lidiar con un largo peregrinaje de consultas y análisis que aumentan la incertidumbre sobre el presente y el futuro de las niñas.
En palabras de las madres y padres: “acceder a un diagnóstico precoz es crucial para nuestras niñas que, gracias a distintos abordajes terapéuticos, pueden mitigar los efectos de la enfermedad en su calidad de vida. El retraso en el desarrollo o la pérdida de habilidades motoras básicas requieren la intervención temprana de un kinesiólogo o fisiatra. Dependiendo de la condición clínica de cada paciente, también son fundamentales la fonoaudiología, la terapia ocupacional, equinoterapia, hidroterapia o musicoterapia. Y desde luego, la consulta permanente a diversos especialistas: un neuropediatra que oriente sobre cuidados diarios y pueda tratar cuadros neurológicos asociados a Rett, como epilepsia o trastornos del sueño, además de traumatólogo-ortopedista, gastroenterólogo, neumólogo entre otros profesionales”.
Objetivo general
Teniendo en cuenta el problema con el que se encuentran los pacientes afectados por el síndrome de Rett y las evidencias experimentales detalladas el proyecto sostiene como objetivo general establecer biomarcadores de diagnóstico del síndrome de Rett y de pronóstico de la severidad de las manifestaciones clínicas de la enfermedad. También, analizar la expresión diferencial de miRNA en vesículas extracelulares derivados de neuronas evaluando los potenciales genes blancos y los procesos biológicos y funciones celulares en los que participan de modo de contribuir a la comprensión de los mecanismos moleculares involucrados en la fisiopatología del síndrome de Rett. Hemos centrado nuestro proyecto en los miRNA considerando que, por su papel en la regulación fina de la expresión génica a nivel postranscripcional, su pleiotropismo y su elevada conservación evolutiva, contribuyen en el mantenimiento y regulación de un gran número de procesos biológicos.
Las participantes
A través de la Fundación tomamos contacto con varias decenas de familias con niñas afectadas por el síndrome de Rett que generosamente aceptaron participar del estudio. Éste se centra en niñas entre 3 y 17 años de edad con un diagnóstico clínico de síndrome de Rett y un análisis genético compatible. Las participantes tienen sus residencias en el área del Gran Buenos Aires y en distintas ciudades del país. Así mismo se recluta un grupo de niñas neurotípicas pareadas por edad y lugar de residencia, de modo de minimizar factores confundentes. El protocolo del proyecto ha sido aprobado por el Comité de Ética de la Sociedad Argentina de Investigación Clínica (SAIC) (código de registro 8033).
Metodología: miRNA como biomarcadores
Luego de ser notificados sobre el proyecto y haber firmado el consentimiento informado, se toman dos muestras de sangre de cada una de las participantes. Una de ellas se utiliza para la extracción de RNA y la evaluación de expresión global de miRNA. La segunda se utiliza para la separación de plasma y obtención de vesículas extracelulares de las que se purificarán aquellas derivadas de neuronas. Junto con los familiares de las niñas se completa una planilla con una serie de datos sociodemográficos y de salud.
Se solicita a los médicos que atienden a cada una de las niñas afectadas por el síndrome de Rett que evalúen la severidad de los síntomas clínicos utilizando una combinación de dos escalas cuantitativas: la Escala de Severidad Clínica y el Análisis de la Conducta Motora [35, 36]. La primera se aplica como una medida de la gravedad clínica basada en características diagnósticas y de desarrollo claves para el síndrome de Rett, incluida la edad de inicio de la regresión, las estereotipias de las manos y el grado de desaceleración adquirida del crecimiento de la cabeza. La segunda se centra en examinar la disfunción motora, conductual y respiratoria. A partir de ambas se construyó una escala de severidad de manifestaciones clínicas (CSS).
Los miRNA que, tras el análisis de expresión y posterior validación, se encuentran expresados diferencialmente entre el grupo de niñas afectadas por el síndrome de Rett y las del grupo neurotípico, y cuya expresión diferencial sea independiente de la edad, tienen un valor potencial como marcadores diagnósticos del síndrome de Rett. Adicionalmente, el análisis de correlación entre cada miRNA expresado diferencialmente y la gravedad de las manifestaciones clínicas según la evaluación de la CSS se utilizará como marcador pronóstico de la gravedad de la enfermedad.
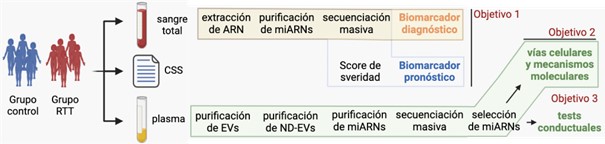
Figura 2: Diseño experimental del proyecto de investigación.
Metodología: miRNA y mecanismos moleculares
La única posibilidad no invasiva de determinar los niveles de expresión de miRNA es en fluidos biológicos como la sangre o la saliva. Si bien MECP2 cumple un rol importante en diversos tejidos y las alteraciones que causa en ellos por la pérdida de su función no pueden ser ignoradas, es en el cerebro donde desempeña un papel fundamental que se refleja en la gravedad de las manifestaciones clínicas que tienen su origen en ese órgano. En este contexto, es probable que los miRNA contenidos en las vesículas extracelulares derivadas de neuronas (ND-EV) reflejen con mayor exactitud lo que ocurre en el cerebro.
Las vesículas extracelulares son liberadas por muchos tipos de células, incluidas las neuronas y las células gliales. Se sugiere que tienen un papel destacado en los trastornos neurológicos. Contienen una variedad de moléculas que incluyen DNA, mRNA, miRNA y proteínas, que pueden transferirse a células vecinas y distantes a través de la circulación, causando cambios fenotípicos en las células receptoras [37, 38]. La transferencia de su contenido, especialmente miRNA, estaría involucrada en varios procesos patológicos [38, 39]. Dado que se ha descubierto que la plasticidad sináptica regula la liberación de ND-EV y que pueden cruzar fácilmente la barrera hematoencefálica, se sugiere que las ND-EV circulantes serían un modelo valioso para comprender la etiología de las enfermedades con base neurológica.
A partir de los miRNA diferencialmente expresados en las ND-EV se predecirán, utilizando algoritmos bioinformáticos específicos, los genes potencialmente alterados por la desregulación de esos miRNA [40, 41]. Luego, se utilizará esta lista de mRNA blancos de los miRNA diferencialmente expresados, para realizar un análisis de enriquecimiento de los términos de ontología génica respecto a procesos biológicos y funciones celulares. Estos análisis permitirán revelar posibles vías celulares alteradas por el síndrome de Rett e iluminar potenciales sitios de intervención terapéutica.
Observaciones finales
El síndrome de Rett es un desorden progresivo del desarrollo neurológico poco frecuente que afecta principalmente a las mujeres. Este trastorno, luego de un desarrollo típico durante los primeros 6 a 18 meses, provoca la pérdida progresiva de las capacidades motoras y del habla. La mayoría de los casos de síndrome de Rett son causados por mutaciones identificables del gen MECP2 en el cromosoma X y pueden presentarse con una amplia gama de discapacidades que van desde leves a graves.
En el estado actual de las investigaciones sobre el Síndrome de Rett hay tres vías de trabajo fundamentales. Una de ellas, por supuesto, la búsqueda de un cura para la enfermedad, la segunda, el diseño de terapias adecuadas para paliar los síntomas de la enfermedad y mejorar la calidad de vida de los pacientes, y la tercera, la búsqueda de un biomarcador que en forma temprana y específica permita diagnosticar la enfermedad y acercar un pronóstico de su progresión. Sin diagnóstico temprano, la implementación de tratamientos oportunos se pierde afectando la calidad de vida del niño y su integración social. En este último punto se encuentra abocado nuestro grupo, con el apoyo y participación de un gran número de niñas afectadas por el síndrome de Rett y sus familiares, y junto con la Fundación SinRett.
Agradecimientos
Los autores quieren agradecer el apoyo y estímulo constante de la Fundación SinRett, especialmente a su presidente el Lic. Fabián Crespo, y a las familias y niñas que participan del proyecto. A la Alianza Argentina de Pacientes (ALAPA) por su apoyo y por el otorgamiento de la beca de investigación a MBC.
Conflicto de intereses
Los autores no tienen conflicto de intereses para declarar.
Referencias:
1. Banerjee A, Miller MT, Li K, Sur M, and Kaufmann WE (2019) Towards a better diagnosis and treatment of Rett syndrome: A model synaptic disorder. Brain. 142, 239–248
2. Vashi N and Justice MJ (2019) Treating Rett syndrome: from mouse models to human therapies. Mammalian Genome. 30, 90–110
3. SchüleB, Armstrong DD, Vogel H, Oviedo A, Francke U (2008) Severe congenital encephalopathy caused by MECP2 null mutations in males: central hypoxia and reduced neuronal dendritic structure. Clinical genetics. 74, 116–126
4. Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, Leonard H, Bailey MES, Schanen NC, Zappella M, Renieri A, Huppke P, Percy AK (2010) Rett syndrome: Revised diagnostic criteria and nomenclature. Annals of Neurology. 68, 944–950
5. Sajan SA, Jhangiani SN, Muzny DM, Gibbs RA, Lupski JR, Glaze DG, Kaufmann WE, Skinner SA, Annese F, Friez MJ, Lane J, Percy AK, Neul JL (2017) Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet Med. 19, 13–19
6. Einspieler C, Marschik PB (2019) Regression in Rett syndrome: Developmental pathways to its onset. Neurosci Biobehav Rev. 98, 320–332
7. Ghosh RP, Nikitina T, Horowitz-Scherer RA, Gierasch LM, Uversky VN, Hite K, Hansen JC, Woodcock CL (2010) Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry. 49, 4395–4410
8. Gold WA, Krishnarajy R, Ellaway C, Christodoulou J (2018) Rett Syndrome: A Genetic Update and Clinical Review Focusing on Comorbidities. ACS Chemical Neuroscience. 9, 167–176
9. Neul JL, Glaze DG, Percy AK, Feyma T, Beisang A, Dinh T, Suter B, Anagnostou E, Snape M, Horrigan J, Jones NE (2015) Improving Treatment Trial Outcomes for Rett Syndrome: The Development of Rett-specific Anchors for the Clinical Global Impression Scale. Journal of child neurology. 30, 1743–1748
10. Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO, Zoghbi H, Percy A, Glaze DG (2008) Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome. Neurology. 70, 1313–1321
11. Kyle SM, Vashi N, Justice MJ (2018) Rett syndrome: A neurological disorder with metabolic components. Open Biology. 10.1098/rsob.170216
12. Cuddapah VA, Pillai RB, Shekar KV, Lane JB, Motil KJ, Skinner SA, Tarquinio DC, Glaze DG, McGwin G, Kaufmann WE, Percy AK, Neul JL, Olsen ML (2014) Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 51, 152–158
13. Caffarelli C, Gonnelli S, Pitinca MDT, Camarri S, Al Refaie A, Hayek J, Nuti R (2020) Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with bone disease severity in Rett syndrome. BMC Med Genet. 21, 21
14. Guy J, Gan J, Selfridge,J, Cobb S, Bird A (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science. 315, 1143–1147
15. Greenberg MVC, Bourc’his D (2019) The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 20, 590–607
16. Tessarz P, Kouzarides T (2014) Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 15, 703–708
17. Marmorstein R, Zhou M-M (2014) Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol. 6, a018762
18. Lyst MJ, Bird A (2015) Rett syndrome: a complex disorder with simple roots. Nature reviews. Genetics. 16, 261–274
19. Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH, Yu M, He C, Sanguinetti G, Sowers LC, Walkinshaw MD, Bird A (2017) MECP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 13, e1006793
20. Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, Rappsilber J, Greenberg ME, Bird A (2013) Rett syndrome mutations abolish the interaction of MECP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 16, 898–902
21. Nott A, Cheng J, Gao F, Lin Y-T, Gjoneska E, Ko T, Minhas P, Zamudio AV, Meng J, Zhang F, Jin P, Tsai L-H (2016) Histone deacetylase 3 associates with MECP2 to regulate FOXO and social behavior. Nat Neurosci. 19, 1497–1505
22. Ebert DH, Gabel HW, Robinson ND, Kastan NR, Hu LS, Cohen S, Navarro AJ, Lyst MJ, Ekiert R, Bird AP, Greenberg ME (2013) Activity-dependent phosphorylation of MECP2 threonine 308 regulates interaction with NCoR. Nature. 499, 341–345
23. Trendel J, Schwarzl T, Horos R, Prakash A, Bateman A, Hentze MW, Krijgsveld J (2019) The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell. 176, 391-403.e19
24. Good KV, Vincent JB, Ausió J (2021) MECP2: The Genetic Driver of Rett Syndrome Epigenetics. Frontiers in Genetics. 10.3389/fgene.2021.620859
25. Chong PA, Vernon RM, Forman-Kay JD (2018) RGG/RG Motif Regions in RNA Binding and Phase Separation. J Mol Biol. 430, 4650–4665
26. Long SW, Ooi JYY, Yau PM, Jones PL (2011) A brain-derived MECP2 complex supports a role for MECP2 in RNA processing. Biosci Rep. 31, 333–343
27. Li R, Dong Q, Yuan X, Zeng X, Gao Y, Chiao C, Li H, Zhao X, Keles S, Wang Z, Chang Q (2016) Misregulation of Alternative Splicing in a Mouse Model of Rett Syndrome. PLoS Genet. 12, e1006129
28. Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 15, 509–524
29. Sheinerman K, Djukic A, Tsivinsky VG, Umansky SR (2019) Brain-enriched microRNAs circulating in plasma as novel biomarkers for Rett syndrome. PLoS ONE. 10.1371/journal.pone.0218623
30. Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, Rosen B, Rodriguez BA, Crawford B, Swaminathan R, Chou S, Li Y, Ziats M, Ernst C, Jaenisch R, Haggarty SJ, Sur M (2018) MECP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Molecular Psychiatry. 23, 1051–1065
31. Conrad T, Marsico A, Gehre M, Orom UA (2014) Microprocessor activity controls differential miRNA biogenesis In Vivo. Cell Rep. 9, 542–554
32. Horvath PM, Piazza MK, Kavalali ET, Monteggia LM (2022) MECP2 loss-of-function dysregulates microRNAs regionally and disrupts excitatory/inhibitory synaptic transmission balance. Hippocampus. 32, 610–623
33. Leonard H, Cobb S, Downs J (2016) Clinical and biological progress over 50 years in Rett syndrome. Nature Reviews Neurology. 10.1038/nrneurol.2016.186
34. Downs J, Rodger J, Li C, Tan X, Hu N, Wong K, De Klerk N, Leonard H (2018) Environmental enrichment intervention for Rett syndrome: An individually randomised stepped wedge trial. Orphanet Journal of Rare Diseases. 10.1186/s13023-017-0752-8
35. Lane JB, Lee HS, Smith LW, Cheng P, Percy AK, Glaze DG, Neul JL, Motil KJ, Barrish JO, Skinner SA, Annese F, McNair L, Graham J, Khwaja O, Barnes K, Krischer JP (2011) Clinical severity and quality of life in children and adolescents with Rett syndrome. Neurology. 77, 1812
36. Ammanuel S, Chan WC, Adler DA, Lakshamanan BM, Gupta SS, Ewen JB, Johnston MV, Marcus CL, Naidu S, Kadam SD (2015) Heightened delta power during slow-wave- sleep in patients with rett syndrome associated with poor sleep efficiency. PLoS ONE. 10.1371/journal.pone.0138113
37. Liu W, Bai X, Zhang A, Huang J, Xu S, Zhang J (2019) Role of Exosomes in Central Nervous System Diseases. Frontiers in Molecular Neuroscience. 12, 1–13
38. Saeedi S, Israel S, Nagy C, Turecki G (2019) The emerging role of exosomes in mental disorders. Translational Psychiatry. 10.1038/s41398-019-0459-9
39. Wei ZX, Xie GJ, Mao X, Zou XP, Liao YJ, Liu QS, Wang H, Cheng Y (2020) Exosomes from patients with major depression cause depressive-like behaviors in mice with involvement of miR-139-5p-regulated neurogenesis. Neuropsychopharmacology. 45, 1050–1058
40. Sticht C, Torre CDL, Parveen A, Gretz N (2018) miRWalk: An online resource for prediction of microRNA binding sites. PLOS ONE. 13, e0206239
41. Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, Chang W (2022) DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221
|
Revista QuímicaViva Número 2, año 23, Agosto 2024 quimicaviva@qb.fcen.uba.ar |