Porfiria aguda intermitente: Presentación de caso
Laura Sabina Varela1, Viviana Alicia Melito1,2, Marcelo Guolo1, Fabiana Alejandra Caballero1, Verónica Goñi1, María del Carmen Martínez1,2, María Pasman3, Valeria Paccioli3, Lucía Tomassi3, Ana María Buzaleh1,2, Victoria Estela Parera1
1 Centro de Investigaciones sobre Porfirinas y Porfirias (CIPYP), UBA-CONICET, Hospital de Clínicas José de San Martín, Buenos Aires, Argentina.
2 Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires, Argentina
3 Hospital General de Agudos J. M. Ramos Mejía. División B. Clínica Médica. Buenos Aires, Argentina
Recibido: 16/07/2024 - Aceptado: 19/08/2024
Resumen
Las Porfirias son enfermedades poco frecuentes consideradas raras o huérfanas causadas por alteraciones en alguna de las enzimas de la biosíntesis del hemo. La Porfiria Aguda Intermitente (PAI) es la más común de las Porfirias agudas y surge como consecuencia de un defecto en el gen HMBS que codifica para la porfobilinógeno deaminasa, tercera enzima del camino biosintético del Hemo. En este trabajo se presenta un caso de una familia con PAI en la cual se han estudiado además del propósito, otros 6 miembros. El diagnóstico diferencial fue fundamental para aplicar el tratamiento específico. Además, en esta familia se pudieron diagnosticar dos familiares que habían sufrido varias crisis sin diagnóstico. El estudio genético fue relevante para identificar un miembro no sintomático entre los familiares y para su asesoramiento evitando así la exposición a factores desencadenantes de la Porfiria.
Palabras clave: Porfirias, Porfiria-Aguda-Intermitente, caso-clínico
Summary
Porphyrias are rare conditions considered orphan diseases, caused by alterations in one of the enzymes of heme biosyntheric pathway. Acute Intermittent Porphyria (AIP) is the most common of acute Porphyrias. It arises as a result of a defect in the HMBS gene that encodes for porphobilinogen deaminase, the third enzyme in the heme biosynthetic pathway. This work presents a case of a family with AIP in which, besides the proband, 6 other members have been studied. Differential diagnosis was crucial in applying the specific treatment. In addition, in this family, two relatives who had suffered several seizures without diagnosis could be diagnosed. The genetic study was relevant for identifying a non-symptomatic member among the relatives and for their counselling, thus avoiding exposure to Porphyria-triggering compounds.
Keywords: Porphyrias, Acute-Intermittent-Porphyria, case-report
Introducción
Las Porfirias son enfermedades poco frecuentes consideradas raras o huérfanas dado que su prevalencia en Argentina y en el mundo oscila entre 1:20.000 y 1:2.350.000 dependiendo del tipo de Porfiria [1-3].
Existen 8 tipos de Porfiria, cada una asociada a la deficiencia parcial, genética o adquirida, de una de las enzimas del camino biosintético del hemo. Como consecuencia del bloqueo de un paso específico de esta secuencia metabólica, en cada Porfiria se produce la acumulación y excreción aumentada de un patrón característico de intermediarios, responsables de la sintomatología presente en cada una de ellas [4-6].
De acuerdo con su sintomatología clínica se clasifican en cutáneas, agudas o mixtas y según el órgano principal de manifestación de la deficiencia enzimática en hepáticas, eritropoyéticas y hepatoeritropoyéticas [4]. De las 8 Porfirias posibles, 4 son cutáneas: Porfiria Cutánea Tardía (PCT), Protoporfiria Eritropoyética (PPE), Porfiria Congénita Eritropoyética (PCE) y Porfiria Hepatoeritropoyética (PHE); 2 agudas: Porfiria Aguda Intermitente (PAI) y Nueva Porfiria Aguda (NPA); 2 mixtas: Porfiria Variegata (PV) y Coproporfiria Hereditaria (CPH)
El defecto enzimático en cada Porfiria es hereditario, aunque la PCT también puede presentarse de forma adquirida. A excepción de la PCE, PHE y NPA, todas se heredan en forma autosómica dominante, con penetrancia incompleta y, en general, los portadores del gen afectado pueden permanecer asintomáticos durante toda su vida. La herencia de un alelo mutado disminuye la actividad de la enzima en aproximadamente un 50%, sin embargo, la cantidad de hemo sintetizado es suficiente para el normal funcionamiento del metabolismo celular [4, 7].
Las Porfirias hepáticas son consideradas enfermedades multifactoriales ya que sólo la mutación en el gen que codifica la enzima deficiente no es un factor determinante para su expresión clínica. Son patologías toxicogenéticas dado que necesitan factores endógenos o exógenos para que el individuo portador presente síntomas. Es posible que la susceptibilidad individual a manifestar la Porfiria en un individuo que heredó la mutación esté determinada por variantes genéticas en otros genes relacionados con la respuesta a los factores desencadenantes.
En el Centro de Investigaciones sobre Porfirinas y Porfirias (CIPYP), a la fecha se han diagnosticado 2282 pacientes con PCT, 196 familias con PAI, 76 familias con PV, 47 familias con PPE, 20 familias con CPH, 5 familias con PCE y 3 familias con PHE que es la forma homocigota de la PCT. También se diagnosticaron 7 familias con Porfiria dual: 4 PAI/PCT, 3 PV/PCT. Cabe destacar que, por lo general, en cada familia hay más de un paciente sintomático.
Las Porfirias agudas se caracterizan por la presencia de severos ataques con sintomatología abdominal aguda y un síndrome neurológico psíquico con crisis intermitentes como consecuencia de la acumulación de precursores, especialmente del ácido 5-aminolevúlico (ALA) que se comporta como una neurotoxina [8, 9].
Los ataques agudos pueden ser precipitados por factores endógenos o exógenos que estimulan la vía de síntesis del hemo ya sea por disminución de los niveles existentes del mismo o por aumento de su requerimiento [4, 5]. La inducción de la biosíntesis, mediante el aumento de actividad de la primera enzima 5-Aminolevúlico sintetasa (ALA-S), junto con la deficiencia en otra de las enzimas de dicha vía, produce la acumulación de precursores desencadenando así la sintomatología característica. Entre los factores endógenos son de relevancia el ayuno, las hormonas esteroides, las infecciones y el estrés; mientras que entre los factores exógenos se encuentran el alcohol, las drogas de abuso y principalmente algunas de uso terapéutico que son administradas para tratar la sintomatología que el paciente refiere antes de sospechar que puede ser una Porfiria. Por esta razón, existen listas de drogas permitidas y no permitidas para los pacientes con Porfirias Agudas (cipypclinicas.blogspot.com, www.drugs-porphyria.org).
Alrededor del 3-5% de los pacientes sufren de ataques agudos graves y recurrentes, que resultan en un marcado deterioro de la calidad de vida. Las complicaciones en la PAI incluyen dolor crónico, falla renal crónica y carcinoma hepatocelular [9-11].
El objetivo del tratamiento de los ataques agudos es reducir el ALA-S hepática, lo que resulta en una disminución de la producción de ALA y PBG. La ingesta o infusión de hidratos de carbono, en cantidades relativamente altas constituye la base del tratamiento nutricional tanto en las crisis como en los períodos asintomáticos ya que actúan inhibiendo el metabolismo de las porfirinas y previniendo la inducción del ALA–S [12]. La terapia con hemina (Normosang) inhibe la síntesis del hemo por retroalimentación negativa sobre ALA-S [13]. Entre las terapias emergentes, se encuentra el tratamiento con RNA de interferencia (RNAi) [13-15]. Específicamente el Givosiran, se utiliza para reducir la expresión del gen ALAS, que codifica para la enzima responsable de la síntesis de ALA. A diferencia de la hemina, que se usa durante los ataques agudos, Givosiran se administra para reducir la frecuencia de los ataques para quienes los tratamientos convencionales no fueron suficientes para controlar el número de crisis.
La PAI es la más común de las Porfirias agudas y la primera en frecuencia en la población argentina (1:150.000) [16]. Surge como consecuencia de un defecto en el gen HMBS que codifica para la porfobilinógeno deaminasa (PBG-D), tercera enzima del camino biosintético del Hemo. Se hereda en forma autosómica dominante y la expresión clínica ocurre generalmente después de la pubertad, aunque hay casos descriptos de PAI infantil (17, 18). A nivel bioquímico, los individuos sintomáticos presentan niveles aumentados de ALA y PBG en orina, responsables de los signos agudos cuya manifestación clínica incluye sintomatología abdominal aguda y un síndrome neurológico psíquico [8, 19].
Hasta la fecha se identificaron más de 553 mutaciones diferentes en el gen de la PBG-D (Human Genome Mutation Database, http://www.hgmd.org) que muestran la heterogeneidad molecular de la PAI. En el CIPYP se han estudiado hasta el momento 129 familias a nivel molecular, de 196 diagnosticadas bioquímicamente. Hemos detectado 19 mutaciones de las cuales la mayoría no habían sido descritas en la literatura antes de ser reportadas por nuestro grupo en estudios previos [8, 16, 20-25]. De estas 19 mutaciones, 1 es una pequeña deleción, 6 causan un cambio de aminoácido, 3 afectan el splicing, 1 es una deleción grande, 7 producen un corrimiento en el marco de lectura que genera una proteína truncada y 1 es una deleción en el marco de lectura que también produce una proteína alterada. Estos hallazgos subrayan la heterogeneidad molecular de la PAI. Sin embargo, el 50% de las familias PAI no relacionadas que fueron analizadas poseen la misma mutación (p.G111R) sugiriendo que tendrían un ancestro común [18].
El objetivo de este trabajo es presentar un caso de una familia con PAI en la cual se han estudiado además del propósito, otros 6 miembros.
Caso clínico
Caso 1 (Propósito)
En julio de 2021, derivan muestras al CIPYP de un Hospital de una provincia argentina de una paciente femenina de 31 años internada con una posible crisis de Porfiria aguda. Tras el análisis se confirmó, tanto bioquímica como genéticamente, que se trataba de una PAI,
Un año antes la paciente había sido hospitalizada en 3 ocasiones debido a dolores abdominales agudos, vómitos, paresia de miembros inferiores e irritabilidad, síntomas que aparecían antes del período menstrual. A pesar de realizarle numerosos estudios se daba el alta sin diagnóstico, indicando tratamiento psiquiátrico. Es probable que la Porfiria se haya desencadenado por el uso de anticonceptivos orales y la ingesta de alcohol.
En la Figura 1 se muestra la evolución de los parámetros bioquímicos de la paciente hasta la fecha indicando los tratamientos recibidos.
Al diagnóstico la paciente fue tratada con una solución de dextrosa al 25% (250 g) con un máximo de 1000 ml por día complementada con ácido fólico (60 a 90 mg/día en tres dosis iguales) y complejo vitamínico B completo. En enero de 2024, ante una nueva crisis, la paciente recibió tratamiento con hemina (Normosang) en una dosis de 3 mg/kg peso al día, durante 4 días. La paciente actualmente se encuentra en remisión recibiendo tratamiento de mantenimiento: dextrosa (120 g/día), ácido fólico (60mg/dia en 3 tomas) y complejo vitamínico B (3 pastillas diarias).
Una hermana de esta paciente, que vive en Suecia, había sufrido varios ataques que la llevaron incluso a ser internada en un psiquiátrico en dicho país. Posteriormente al diagnóstico del propósito, fue también diagnosticada con PAI y en 2024 se realizó un control bioquímico en el CIPYP.
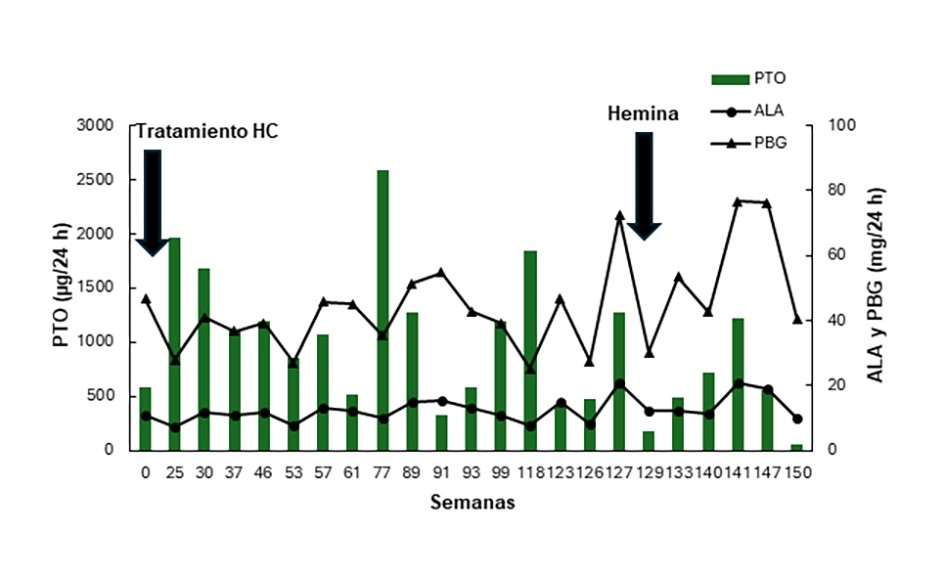
Figura 1: Evolución de los parámetros bioquímicos del propósito. Valores Normales: Ácido 5-aminolévulico (ALA): ≤4 mg/24 h. Porfobilinógeno (PBG): ≤2 mg/24 h. Porfirinas totales en orina (PTO): 20-250 µg/24 h. HC: hidratos de carbono.
Caso 2
Una prima segunda por parte paterna del propósito fue operada de abdomen agudo por probable endometriosis a los 21 años. A los 30 años, luego de recibir tratamiento con hormona folículo estimulante y estrógenos para inducir la ovulación tuvo una crisis de abdomen agudo médico con 7 días de dolor, pérdida de peso y astenia. A las 4 semanas de gestación de su primer embarazo tuvo otro ataque, estando internada durante un mes.
En diciembre de 2021 presentó sintomatología neuroabdominal, hiponatremia grave y pérdida de conocimiento. Con el antecedente de su prima, se pensó en Porfiria y finalmente se diagnosticó con PAI. Recibió tratamiento con hidratos de carbono estando actualmente en remisión y continuando con la terapia de mantenimiento.
En la Figura 2 se muestra la evolución de los parámetros bioquímicos hasta la fecha de la paciente indicando el tratamiento recibido.
Tiene 2 hijas de 4 y 2 años; una de ellas heredó la variante patogénica familiar (HMBS(NM_000190.4):c.849G>A, (p.Trp283Ter)) y a la fecha está diagnosticada con PAI latente.
Su madre, que no presentaba síntomas, fue diagnosticada también con PAI.
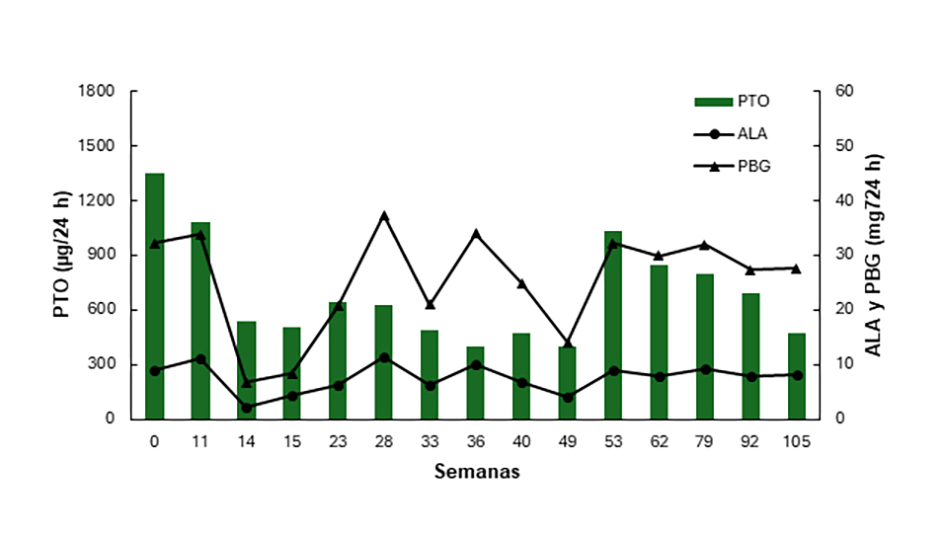
Figura 2: Evolución de los parámetros bioquímicos del caso 2. Valores Normales: Ácido 5-aminolévulico (ALA): ≤4 mg/24 h. Porfobilinógeno (PBG): ≤2 mg/24 h. Porfirinas totales en orina (PTO): 20-250 µg/24 h. HC: hidratos de carbono.
En la Figura 3 se muestra el árbol genealógico de la familia estudiada.
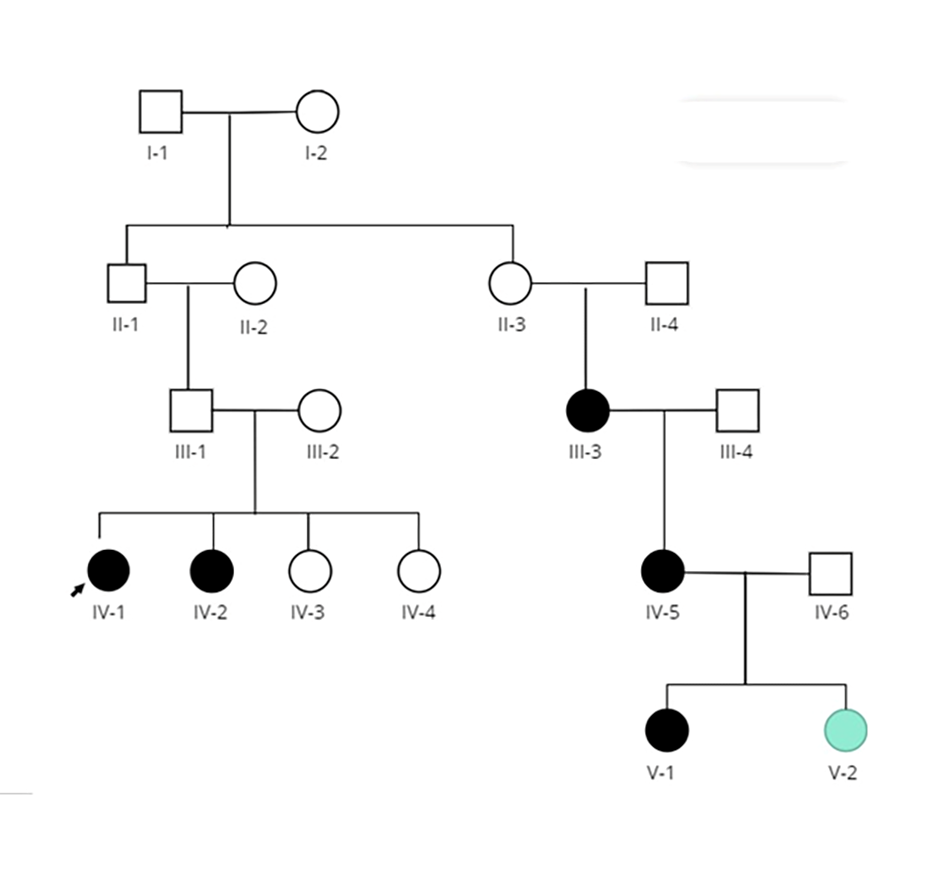
Figura 3: Genealogía de la familia con Porfiria Aguda Intermitente. La flecha señala la paciente femenina que corresponde al propósito. (○, □) No analizados. (●, ■) c.849G>A en heterocigosis. (●) No portador
En la Tabla I se describen los datos bioquímicos del propósito y sus familiares y la mutación en el gen HMBS al momento del diagnóstico.
Tabla I: Datos bioquímicos y genéticos de los individuos estudiados en la familia.
| Paciente | Fecha | Edad (años) | Test de PBG | ALA (mg/24 h) | PBG (mg/24 h) | PTO (µg/24 h) | IPP | PBG-D (U/ml GR) | Mutación en gen HMBS |
|---|---|---|---|---|---|---|---|---|---|
| 1 (propósito) | 22/07/21 | 31 | + | 11,1 | 46,7 | 583 | 1,10 | 41,27 | p.Trp283Ter |
| 2 (prima 2da de 1) | 13/12/21 | 35 | + | 9,0 | 32,2 | 1.325 | 1,25 | 47,44 | p.Trp283Ter |
| 3 (hija de 2) | 28/07/22 | 2 | - | ND | ND | ND | 1,00 | 75,11 | p.Trp283Ter |
| 4 (hija de 2) | 28/07/22 | 4 | - | ND | ND | ND | 1,00 | 81,83 | Normal |
| 5 (madre de 2) | 10/01/22 | 64 | + | 3,6 | 9,5 | 266 | 1,17 | 63,48 | p.Trp283Ter |
| 6 (hermana de 2) | 12/09/22 | 41 | - | 1,3 | 1,6 | 105 | 1,00 | 79,56 | ND |
| 7 (hermana de 1) | 23/02/24 | 42 | + | 3,2 | 8,1 | 241 | ND | ND | p.Trp283Ter |
ND: No determinado. Valores Normales: Ácido 5-aminolévulico (ALA): ≤4 mg/24 h. Porfobilinógeno (PBG): ≤2 mg/24 h. Porfirinas totales en orina (PTO): 20-250 µg/24 h. Índice de porfirinas plasmáticas (IPP): ≤1,30. Porfobilinógeno deaminasa (PBG-D): 81,51±11,96 U/ml GR;
Conclusiones
Las Porfirias son enfermedades raras, poco frecuentes y dada la inespecificidad de su sintomatología en muchos casos no se llega a su diagnóstico rápidamente y se la confunde con otras patologías. Sin embargo, una vez que se sospecha una Porfiria, su diagnóstico en los individuos sintomáticos es relativamente simple y seguro mediante los estudios bioquímicos que permiten determinar qué intermediarios del camino del hemo están aumentados en orina, sangre y/o materia fecal para realizar el diagnóstico diferencial de Porfiria. Este diagnóstico diferencial es fundamental tanto para realizar el tratamiento adecuado al paciente y colaborar con la rápida detección e identificación de la mutación en el gen que codifica la enzima afectada responsable de cada Porfiria que permite la detección de portadores asintomáticos (latentes) en cada familia diagnosticada. El asesoramiento es relevante para evitar la exposición a factores porfirinogénicos a fin de prevenir el desencadenamiento de la Porfiria.
Los desórdenes del metabolismo de las porfirinas tienen una expresión clínica polimorfa que pueden variar desde cuadros de intensa fotosensibilización a una crisis de abdomen agudo asociado a neuropatías, nefropatías o hepatopatías, e incluso causar la muerte en el 10% de los casos en las formas agudas. El fundamento del tratamiento en el ataque agudo, desde el enfoque fisiopatológico, es actuar directamente sobre el camino del hemo y de esta forma controlar los desequilibrios producidos por la acumulación de los intermediarios que llevan al desarrollo del ataque, y paralelamente, identificar y eliminar él o los factores precipitantes.
Cabe destacar que en esta familia luego de estudiar al propósito se pudieron diagnosticar 2 familiares que habían sufrido varias crisis sin que éstas fueran asociadas a una Porfíria y otro familiar asintomático con valores levemente elevados de PBG. Además, se diagnosticó un caso de PAI latente y otro normal que no heredó la variante familiar patogénica.
Debemos remarcar la importancia de que se piense en Porfirias para poder llegar sin dilación a su diagnóstico e implementar rápidamente el tratamiento adecuado, evitando consecuencias graves que incluso pueden llevar a la muerte si se utilizan medicamentos y/o tratamientos quirúrgicos que puedan exacerbar la crisis. El manejo clínico-nutricional de los pacientes con Porfiria asegura la pronta recuperación de la crisis y la prolongación del período de remisión por un largo tiempo y en muchos casos para toda la vida.
Agradecimientos
Se agradece la colaboración de la Sra. Victoria Castillo en la recepción y manipulación de las muestras biológicas de los pacientes que asisten al CIPYP.
Aspectos éticos
Se cumplieron estrictamente con todos los requisitos éticos, legales y jurídicos, establecidos en las normas bioéticas nacionales –Disposición ANMAT 6677/10– e internacionales -Código de Nüremberg, Declaración de Helsinki y sus modificaciones; así como también la Declaración Universal sobre Genoma Humano y de Los Derechos Humanos aprobada por la Conferencia General de la UNESCO del 11/11/97. Se dispone además de la evaluación del Comité Independiente de Ética del CIPYP. En todos los casos se entregó el formulario de Consentimiento Informado.
Declaración de conflicto de intereses: Los autores declaran no poseer conflicto de intereses.
Referencias:
1. Elder G, Harper P, Badminton M, Sandberg S, Deybach JC (2013) The incidence of inherited porphyrias in Europe. Journal of Inherited Metabolism Disease 36: 849-857, doi: 10.1007/s10545-012-9544-4.
2. Ramanujam VS, Anderson KE (2015) Porphyria Diagnostics-Part 1: A Brief Overview of the Porphyrias. Current Protocols in Human Genetics 86: 17.20.1-17.20.26. doi: 10.1002/0471142905.hg1720s86.
3. Ma L, Tian Y, Peng C, Zhang Y, Zhang S (2020). Recent advances in the epidemiology and genetics of acute intermittent porphyria. Intractable & Rare Diseases Research 9(4): 196-204. doi: 10.5582/irdr.2020.03082.
4. Rossetti MV, Buzaleh AM, Parera VE, Fukuda H, Lombardo ME, Lavandera J, Gerez EN, Melito VA, Zuccoli JR, Ruspini, SV, Puente VR, Diez BA, Teijo MJ, Cerbino G, Varela LS, Guolo MN, Batlle A (2016) Acta Bioquímica Clínica Latinoamericana Libro de Oro 50 (4) (2016) 547-573.
5. Phillips JD (2019) Heme biosynthesis and the porphyrias. Molecular Genetic and Metabolism, 28, 164-177, doi:10.1016/j.ymgme.2019.04.008.
6. Dickey AK, Leaf RK, Balwani M (2024) Update on the Porphyrias. Annual Review Medicine 75: 321-335, doi: 10.1146/annurev-med-042921-123602.
7. Yasuda M, Chen B, Desnick RJ (2019) Recent advances on porphyria genetics: Inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Molecular Genetics and Metabolism 128(3): 320-331. doi: 10.1016/j.ymgme.2018.11.012.
8. Martinez M del C, Cerbino GN, Granata BX, Batlle A, Parera VE, Rossetti MV (2021) Clinical, biochemical, and genetic characterization of acute hepatic porphyrias in a cohort of Argentine patients. Molecular Genetics & Genomic Medicine 9: 5.: e1059,. doi:10.1002/mgg3.1059., 9:e1059.
9. Ricci A, Di Pierro E, Marcacci M, Ventura P (2021) Mechanisms of neuronal damage in acute hepatic porphyrias. Diagnostics (Basel) 11(12): 2205. doi: 10.3390/diagnostics11122205.
10. Pallet N, Mami I, Schmitt C, Karim Z, François A, Rabant M, Nochy D, Gouya L, Deybach JC, Xu-Dubois Y, Thervet E, Puy H, Karras A (2015) High prevalence of and potential mechanisms for chronic kidney disease in patients with acute intermittent porphyria. Kidney International 88: 386-395, doi:10.1038/ki.2015.97.
11. Melito V, Varela L, Antinucci F, Brutti J, Maurette R, Tomassi L, Buzaleh A, Parera V, Anders M (2022) Acute Intermittent Porphyria as a risk factor for hepatocellular carcinoma: first case in Argentina. Medicina 82: 194, Res 363.
12. Martin L, Tomassi L, Parera V, Batlle A (2004) Manejo clínico - nutricional de las Porfirias Agudas. Revista del Hospital General de Agudos J. M. Ramos Mejía IX (1). http://www.ramosmejia.org.ar
13. Fontanellas A, Ávila MA, Anderson KE, Deybach JC (2019) Current and innovative emerging therapies for porphyrias with hepatic involvement. Journal of Hepatology 71(2):422-433. doi: 10.1016/j.jhep.2019.05.003.
14. Fontanellas A, Ávila MA, Arranz E, Enríquez de Salamanca R, Morales-Conejo M (2021) Acute intermittent porphyria, givosiran, and homocysteine. Journal of Inherited Metabolic Disease 44(4): 790-791. doi: 10.1002/jimd.12411.
15. Yasuda M, Keel S, Balwani M (2023) RNA interference therapy in acute hepatic porphyrias. Blood 142(19): 1589-1599, doi: 10.1182/blood.2022018662.
16. Parera VE, De Siervi A, Varela L, Rossetti MV, Batlle A (2003) Acute porphyrias in the Argentinean population: a review. Cellular and Molecular Biology. (Noisy-le-grand) 49, 493-500.
17. Melito VA, Rossetti MV, Parera VE, Batlle A (2006) Porfirias poco frecuentes. Casos detectados en la población argentina. Revista Argentina de Dermatología 87: 248-263.
18. Cerbino GN, Gerez EN, Varela LS, Melito VA, Parera VE, Batlle A, Rossetti MV (2015) Acute intermittent porphyria in Argentina: an update. BioMed Research International 2015 (2015): 946387, doi:10.1155/2015/946387.
19. Pischik E, Baumann K, Karpenko A (2023) Pathogenesis of acute encephalopathy in acute hepatic porphyria. Journal of Neurology 270: 2613-2630, doi:10.1007/s00415-023-11586-5.
20. De Siervi A, Mendez M, Parera VE, Varela LS, Batlle A, Rossetti MV (1999) Acute intermittent porphyria: characterization of two novel mutations in the porphobilinogen deaminase gene, one amino acid deletion (453-455delAGC) and one splicing aceptor site mutation (IVS8-1G→T). Human Mutation. Mutation in Brief 14(4): 355, doi:10.1002/(SICI)1098-1004(199910)14:4<355::AID-HUMU19>3.0.CO;2-T.
21. De Siervi A, Rossetti MV, Parera VE, Astrin KH, Aizencang GI, Glass IA, Batlle A, Descnik R (1999) Identification and characterization of Hydroxymethylbilane Synthase mutations causing Acute Intermittent Porphyria: Evidence for an ancestral founder of the common G111R mutation. American Journal of Medical Genetics 86: 366-375.
22. De Siervi A, Rossetti MV, Parera VE, Mendez M, Varela LS, Batlle AM del C (1999) Acute intermittent porphyria: Biochemical and clinical analysis in the Argentinean population. Clinica Chimica Acta 288: 63-71, doi:10.1016/s0009-8981(99)00139-4
23. De Siervi A, Weiss Cádiz DE, Parera VE, Batlle A, Rossetti MV (2000) Identification and Characterization of two novel mutations that produce Acute Intermittent Porphyria: a 3-base deletion (841-843delGGA) and a missense mutation (T35M). Human Mutation Mutation in Brief. 16 (4): 373.
24. Flagel MS, Parera VE, Batlle A, Rossetti MV (2011) A novel duplication mutation in Argentinean Acute Intermittent Porphyria patients. Gene Bank HQ731552.
25. Flagel MS, Parera VE, Batlle A, Rossetti MV (2012) Caracterización de una deleción intrónica en el gen de la PBGD para el diagnóstico presiintomático en Porfiria Aguda Intermitente [resumen]. Medicina 72 (Supl II): 234.
|
Revista QuímicaViva Número 2, año 23, Agosto 2024 quimicaviva@qb.fcen.uba.ar |