CUATRO AÑOS DE PAMPA:
DIAGNÓSTICO MOLECULAR DE ENFERMEDADES AUTOINFLAMATORIAS EN ARGENTINA
Guadalupe Buda2, Lorenzo Erra2, Silvia Daniellian3, Graciela Espada4, Matías Oleastro3, Ana Clara Lugones1, Marcelo A. Martí2*
1. BITGENIA, Alicia Moreau de Justo 1750 Piso 3H Ciudad Autónoma de Buenos Aires, Argentina
2. Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires (FCEyN-UBA) e Instituto de Química Biológica de la Facultad de Ciencias Exactas y Naturales (IQUIBICEN) CONICET, Pabellón 2 de Ciudad Universitaria, Ciudad de Buenos Aires C1428EHA, Argentina.
3. Hospital Juan P. Garrahan, Servicio de Inmunología y Reumatología, Buenos Aires, Argentina
4. Hospital de Niños Dr. Ricardo Gutiérrez, Servicio de Reumatología, Buenos Aires, Argentina.
Recibido: 11/04/2022 - Aceptado: 27/06/2022
Resumen
Las enfermedades autoinflamatorias engloban un conjunto de Enfermedades Poco Frecuentes caracterizadas todas ellas por la presencia de inflamación sistémica crónica o recurrente, como consecuencia de una disregulación del control del proceso inflamatorio. Muchas son hereditarias y monogénicas. Su variabilidad clínica y el elevado número de genes asociados a las mismas, favorece su abordaje por técnicas de secuenciación masiva.
En este contexto, nació en el año 2018 el Programa Argentino de Medicina de Precisión en enfermedades Autoinflamatorias, con el apoyo de Novartis y el acompañamiento de un consejo asesor, compuesto por médicos y bioquímicos especializados en Genética, Inmunología y Reumatología. El proyecto tuvo 3 Ediciones, incorporó a más de 160 pacientes y tuvo como principal objetivo brindarles a los pacientes la posibilidad de acceder a un diagnóstico preciso de forma gratuita. Los resultados muestran que la tasa de éxito -casos donde se arribó a un diagnóstico molecular– se estimó entre un 15-35% lo que es consistente con estudios realizados a nivel internacional. Varios de los casos, además, han permitido encontrar variantes nóveles que representan el punto de partida para el estudio de los fenómenos moleculares subyacentes al desarrollo patológico.
La exitosa implementación y desarrollo de tres Ediciones del Proyecto PAMPA durante cuatro años consecutivos, constituye una clara evidencia de que es posible implementar servicios de diagnóstico molecular basados en secuenciación de próxima generación a nivel nacional.
Palabras clave: NGS, Exoma, Enfermedades Autoinflamatorias, Proyecto PAMPA.
FOUR YEARS OF PAMPA: MOLECULAR DIAGNOSIS OF AUTOINFLAMMATORY DISEASES MADE IN ARGENTINA
Summary
Systemic autoinflammatory diseases encompass a group of Rare Diseases characterized by the presence of chronic or recurrent systemic inflammation, as a consequence of inflammatory pathways dysregulation. Many are hereditary and monogenic. Their clinical variability and the high number of associated genes makes this group of disorders an ideal case for massive sequencing technologies.
In this context, the Argentine Precision Medicine Program in Autoinflammatory diseases, better known as PAMPA -(for its Spanish acronym, https://apps.bitgenia.com/pampa/)-, started at the beginning of 2018, with the support of Novartis and the accompaniment of a Medical Advisory Board. The 3 editions of the project incorporated over 160 patients with suspected AIDs, aiming to provide an accurate molecular diagnosis, bringing an opportunity to receive adequate therapy in the context of a true personalized medicine. The diagnostic efficiency was estimated between 15-35%, which is consistent with previous international studies. Several cases also led to the discovery of novel variants that became a trigger for further molecular studies to research the underlying causes of the pathological process.
The successful development of three PAMPA Editions during four consecutive years constitutes clear evidence that it is possible to implement molecular diagnostic services based on next generation sequencing at a national level.
Keywords: NGS, Exome, Autoinflammatory Diseases, PAMPA Project.
Introducción
El concepto de enfermedad autoinflamatoria sistémica (EAI) fue propuesto en 1999 por el doctor Kastner para agrupar algunas enfermedades poco frecuentes (EpoF) con manifestaciones clínicas similares (episodios febriles e inflamatorios recurrentes), y se estableció que la ausencia de autoanticuerpos a títulos elevados o de células T específicas de antígenos propios, eran características diferenciales respecto de las enfermedades autoinmunes [1-3]. Desde entonces, el número de enfermedades autoinflamatorias hereditarias ha ido aumentando progresivamente, debido principalmente a un creciente conocimiento del funcionamiento del sistema inmune, y a los avances en genética humana.
En la tabla I se incluyen algunos ejemplos de EAI poligénicas o multifactoriales y se las distingue de las formas monogénicas, divididas en tres grupos: Infammosomopatias, Interferonopatias y Relopatias. Adicionalmente, se las clasifica de una manera gráfica teniendo en cuenta la vía afectada, sus manifestaciones clínicas más destacadas, y el target terapéutico [4–8] (Ver Tabla I).
| Monogénicas | Poligénicas | |||
|---|---|---|---|---|
| Inflamasomopatías | Interferonopatías | Relopatías | Otras |
|
|
|
|
| |
| Vía afectada | ||||
| IL-1 | IFN tipo I | NF-kB, IL-1, IL-6, TNF | ||
| Clínica y tratamiento | ||||
| Fiebre y rash Distinto espectro de severidad | Calcificación CNS, vasculopatía y fibrosis acral y pulmonar | Granuloma, Uveítis, Úlceras | ||
| Sobreactivación del inflamasoma | Respuesta aumentada a IFN | Ubiquitilación anormal | ||
| Tratamiento: Inhibidores de IL-1 | Tratamiento: Inhibidores JAK | Tratamiento: Anti-TNF | ||
Tabla I: Enfermedades autoinflamatorias monogénicas y poligénicas: clasificación, vía afectada, manifestaciones clínicas características y target terapéutico [5–9].
FMF, Fiebre Mediterránea Familiar; MKD/HIDS, Deficiencia de mevalonato quinasa (Síndrome de hiper-IgD); TRAPS, Síndrome Periódico Asociado al Receptor del Factor de Necrosis Tumoral; CAPS, Síndrome de Fiebre Periódica Asociada a Criopirina; PAPA, Síndrome de artritis piógena estéril, pioderma gangrenoso y acné; SAVI, Vasculopatía asociada a STING con inicio en la infancia; Síndrome de dermatosis neutrofílica crónica atípica con lipodistrofia y temperatura elevada; HA20, Haploinsuficiencia A20; DADA2, Deficiencia de la adenosina desaminasa-2; DIRA, Deficiencia del antagonista del receptor de la Interleucina 1; DITRA, Deficiencia del antagonista del receptor de la Interleucina-36; PFAPA, Fiebre periódica asociada a adenitis, faringitis y estomatitis aftosa; CNO/CRMO, Osteomielitis crónica no bacteriana/osteomielitis crónica multifocal recurrente; AIJs, Artritis idiopática juvenil sistémica; SAPHO, Sinovitis, Acné, Pustulosis, Hiperostosis y Osteítis; NF-kB, Factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas; TNF, Factor de necrosis tumoral, IFN: Interferón, IL: interleucina.
Como se muestra en la Tabla I y según su origen, las enfermedades autoinflamatorias se pueden dividir en [9]:
- Enfermedades autoinflamatorias monogénicas: son aquellas causadas por mutaciones en genes que codifican proteínas implicadas en vías inflamatorias, cuya alteración conduce a una respuesta inflamatoria sistémica exagerada por la producción descontrolada de citocinas proinflamatorias, principalmente la IL-1β, en el caso de las inflamasomopatías (FMF, TRAPs, MKD y CAPS). La mayoría son heredadas y sigue un patrón de herencia mendeliano dominante o recesivo, aunque algunas enfermedades se han descrito asociadas a mutaciones de novo. Generalmente, se presentan durante la infancia o adolescencia, pero en algunas ocasiones alcanzan la edad adulta debido al retraso diagnóstico. Se caracterizan por episodios de inflamación sistémica órgano-específica, con un amplio espectro de las características inflamatorias.
- Enfermedades autoinflamatorias poligénicas o multifactoriales: son enfermedades que presentan una base autoinflamatoria pero etiología genética desconocida [10]. Algunas de ellas están mediadas por la IL-1, ya que responden a su bloqueo específico. Entre éstas se encuentran el síndrome de PFAPA [11], Osteomielitis crónica no bacteriana/osteomielitis crónica multifocal recurrente (CRMO/CNO), la Enfermedad de Crohn, la Artritis Idiopática Juvenil Sistémica (AIJ sistémica), la enfermedad de Still del adulto, el Síndrome de Schnitzler [2,10],el Síndrome SAPHO (acrónimo de sinovitis, acné, pustulosis, hiperostosis y osteítis) y la gota, entre otras.
- Enfermedades autoinflamatorias indiferenciadas (o indefinidas): corresponden a cuadros o síndromes clínicos que cursan con fiebre recurrente o persistente, normalmente junto a otras manifestaciones inflamatorias, en los que se han descartado las causas infecciosas, neoplásicas y autoinmunes, y el estudio genético es negativo o no concluyente (p.ej. presencia de variantes benignas o de penetrancia incompleta, no asociadas con la expresión clínica). Además, suelen responder a fármacos utilizados en las enfermedades monogénicas, como la colchicina, dosis altas de glucocorticoides y/o agentes anti-IL-1. Actualmente, se reconoce que un fenotipo autoinflamatorio clásico o atípico, también puede ser ocasionado por mutaciones determinantes de mosaicismos somáticos, gonadales o gonosomales [11,12]. En este contexto, los pacientes sin mutaciones identificables continúan siendo un enigma y el mosaicismo somático podría proveer una explicación a algunos de estos dilemas clínicos, generando nuevos desafíos en la comprensión y el tratamiento de las enfermedades de transmisión genética [13].
El diagnóstico de las EAI es clínico y se basa en las características fenotípicas evidenciadas en los pacientes. Las EAI más frecuentes se asocian a fiebre periódica, erupciones cutáneas, compromiso de serosas y elevación de reactantes de fase aguda (RFA). Sin embargo, existe una gran variabilidad clínica, que incluye vasculopatías, enteropatías y, en algunos casos, alteraciones neurológicas [14]. Los autoanticuerpos, aunque usualmente ausentes, pueden observarse en ciertos síndromes.
Actualmente, las tecnologías de secuenciación masiva permiten determinar, en tiempos y a costos accesibles, la secuencia completa de nuestros genes dándonos la oportunidad de precisar cuál (o cuáles) son las variantes responsables del desarrollo patológico en un porcentaje moderado de los casos. El diagnóstico genético es muy importante, pero debe ser realizado e interpretado correctamente. Por lo tanto, es necesario contar con un equipo profesional con acceso a tecnologías adecuadas y con la experiencia suficiente acumulada en la interpretación y validación de variantes de significado incierto y de nuevas mutaciones, que se detectan con mucha frecuencia. El correcto diagnóstico, permite en muchos casos un abordaje terapéutico con agentes biológicos que bloquean citocinas proinflamatorias, particularmente IL-1 [15], anti-TNF [16] y bloqueantes de IFN [17,18], que han demostrado ser efectivos en muchos pacientes.
En el año 2018 surgió el proyecto PAMPA, que tuvo tres Ediciones, incorporó a más de 160 pacientes con sospecha de EAI y buscó brindarles la posibilidad de acceder a un diagnóstico preciso de forma gratuita, promoviendo de esta manera la oportunidad de recibir un tratamiento adecuado en el contexto de una verdadera medicina personalizada.
Materiales y Métodos
La difusión del proyecto tuvo un enorme impacto en todo el territorio argentino y permitió la inclusión de pacientes provenientes de instituciones de salud tanto públicas como privadas. La postulación de casos fue realizada a través de un formulario online por diversos profesionales de la salud, especializados en identificar pacientes con sospecha de Enfermedad Autoinflamatoria. La postulación del caso incluye además la firma por parte del paciente del consentimiento informado correspondiente, y la aprobación del mismo por parte del comité de ética de la institución que lo postula (ver sección correspondiente). Como criterio de selección se dio prioridad a aquellos casos en los cuales el profesional médico tuviera sospecha diagnóstica clínica de EAI. Los escenarios clínicos más frecuentes de presentación de estas enfermedades, en función de lo establecido por Federici y Gattorno [19-20], fueron adaptados a las ediciones de PAMPA y contemplaron la inclusión de pacientes con:
- Fiebre recurrente o periódica;
- Fiebre crónica y eczema;
- Formas atípicas de artritis recurrente o crónica;
- Lesiones pustulosas e inflamación sistémica;
- Lesiones osteolíticas inflamatorias estériles;
- Lesiones cutáneas afebriles.
En todos estos escearios, el ejercicio clínico de diagnóstico diferencial con enfermedades infecciosas, inflamatorias autoinmunes, y oncológicas resultó fundamental para enfocar el diagnóstico de estas entidades. Se descartaron aquellos casos con una fuerte sospecha de enfermedad multifactorial o que tuvieran una alta probabilidad de ser causados por modificaciones genéticas no detectables por medio de la secuenciación exómica. Todos los casos postulados fueron analizados en profundidad por los integrantes del Advisory Board y se seleccionaron aquellos que cumplieron con los criterios de inclusión. Todos los casos analizados formaron parte de: i) protocolos investigación aprobados por los comités de ética de las instituciones que atienden los pacientes, con los consentimientos informados correspondientes; o alternativamente ii) casos enmarcados en un proceso de innovación clínica [21] donde el médico responsable del caso otorga y explica el consentimiento informado al paciente. En todos los casos el análisis se limitó a aquellos genes previamente consensuados con el profesional como directamente relacionados con el diagnóstico presuntivo, para disminuir la posibilidad de hallazgos incidentales.
Una vez aprobado el caso, e informados el médico y paciente, se solicitó la firma del consentimiento informado y la declaración jurada. Luego se procedió con la extracción de una muestra de sangre periférica y a la purificación de ADN de linfocitos circulantes en cada caso, mediante la utilización de kits comerciales QIAamp DNA Mini Kit (QIAGEN), logrando una concentración final mínima de 50 ng/ul y una pureza mayor a 1,8 en relación de absorbancia 260nm/280nm.
La captura exómica se realizó con el kit de “Agilent SureSelect Human All Exon V5 (Edición 2018)”, “Agilent SureSelect Human All Exon V7 (Edición 2019-2020)” y las muestras fueron secuenciadas mediante la tecnología “Illumina HiSeq 4000” con una longitud de lectura de 100 pares de bases y una profundidad promedio de 100X. Todas las muestras fueron anonimizadas en todos los pasos del análisis, desde la extracción de sangre hasta la entrega de resultados finales al médico responsable.
Las lecturas pareadas obtenidas fueron alineadas contra el genoma de referencia de “The Genome Reference Consortium” en su versión 37 (GRCh37), por medio del software Barrow Wheelers Aligner (BWA) [22]. El procesamiento de las lecturas alineadas se llevó a cabo según las recomendaciones del Genome Analysis Toolkit (GATK) [23–25]. En primer lugar se procedió a marcar las lecturas que pudieran ser producto de duplicaciones de PCR por medio del software PICARD, se detectaron regiones con varias lecturas de baja calidad y se realizó un realineado local con el fin de detectar posibles deleciones o inserciones pequeñas. La tipificación de haplotipos y la genotipificación conjunta también se realizaron siguiendo la recomendación de GATK, utilizando la herramienta "Haplotype Caller" de GATK. El siguiente paso consistió en detectar las diferencias entre el consenso de las lecturas obtenidas y el genoma de referencia, en un proceso denominado “llamado de variantes” (del inglés “variant calling”) mediante la herramienta “Haplotype Caller” de GATK. A partir de la información de calidad de las variantes se realizó un filtrado, con el protocolo “Hard Filtering”, según las recomendaciones detalladas por GATK, con el fin de detectar y filtrar aquellas variantes llamadas de baja calidad. Como último paso se procedió a anotar el VCF (vincular las variantes con datos biológicos) con información externa de bases de datos utilizando el paquete de software SnpEff/SnpSift [26] y Annovar [27]. Para ello se utilizaron las siguientes fuentes: dbSNP, ExAC, 1000 Genomas, GnomAD para frecuencia poblacional [28], ClinVar [29] para la relevancia clínica y Polyphen [30], SIFT [31] y Mutation Taster [32], entre otros, para las predicciones de patogenicidad.
A partir de la información anotada en las variantes y en base a su impacto molecular, se seleccionaron aquellas con mayor probabilidad de ser causantes del cuadro clínico presentado, en un proceso denominado priorización de variantes utilizando el software B_platform. Para la priorización de variantes se utilizó un panel de genes asociados a EAI, que fueron determinados por el Advisory Board y actualizados anualmente.
El panel principal incluyó, entre otros, los siguientes genes: ACP5, ACTB, ADAM17, ADAR, ADGRE2, AGBL3, AP1S3, ARPC1B, CARD14, CECR1, COPA, DDX58, DNASE2, EGFR, IFIH1, IL10RA, IL10RB, IL1RN, IL36RN, ISG15, LACC1, LPIN2, MEFV, MVK, NCSTN, NLRC4, NLRP1, NLRP12, NLRP3, NLRP7, NOD2, OSMR, OTULIN, PLCG2, POLA1, POMP, PSMA3, PSMB4, PSMB8, PSMB9, PSMG2, PSTPIP1, RBCK1, RELA, RNASEH2A, RNASEH2B, RNASEH2C, RNF31, SAMHD1, SH3BP2, SHARPIN, SLC29A3, STIM1, TMEM173, TNFAIP3, TNFRSF11A, TNFRSF1A, TREX1, TRNT1, USP18, WDR1.
En caso de no encontrar variantes de relevancia clínica en el panel principal, se procedió a analizar un panel ampliado, que contempló el estudio adicional de aquellos genes de interés para cada caso en particular. En general, la búsqueda adicional se concentró en paneles de Inmunodeficiencias primarias, enfermedades autoinmunes, enfermedad inflamatoria intestinal, de acuerdo con la historia clínica del paciente en cuestión y en función de las sugerencias y recomendaciones realizadas por el Advisory Board y la médica genetista del equipo.
Todos los casos fueron sometidos al protocolo de priorización de variantes diseñado previamente, con la finalidad de descartar variantes que no cumplieran con los requisitos para ser consideradas como “patogénicas”, “posiblemente patogénicas” o de “significado incierto” según los criterios del American College of Medical Genetics and Genomics (ACMG) [33] y de enriquecer los resultados con aquellas variantes candidatas a ser las causales de la sintomatología presentada. Dicho protocolo consta de grupos de filtros (descritos en el Figura 1), que se aplican secuencialmente y para los cuales se analizan los resultados de manera detallada.
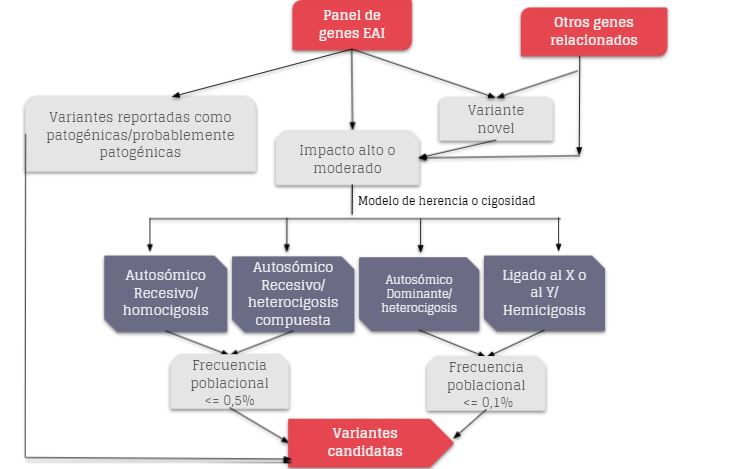
Figura 1: Esquema de priorización de variantes. En primer lugar se buscaron variantes reportadas en bases de datos. Luego, se analizaron variantes de alto impacto y baja frecuencia poblacional, según el modelo de herencia ó nóveles en los genes candidato. De no encontrarse resultados de relevancia, se continuó con la búsqueda de variantes nóveles de impacto alto o moderado en genes indirectamente relacionados a la sospecha clínica.
Brevemente, cada variante fue categorizada primero de acuerdo a su impacto a nivel molecular. Se le asignó un alto impacto a aquellas variantes que resultan en ganancia o pérdida de codones de inicio, finalización de la traducción (stop), cambios en el marco de lectura y/o variantes en los sitios de splicing. El impacto se consideró moderado cuando las modificaciones a nivel proteico involucran cambios no sinónimos de un único aminoácido, y/o pequeñas inserciones o deleciones que conserven el marco de lectura. El resto se consideró de bajo impacto. Luego, para cada variante se analizó su frecuencia poblacional (si la hubiera), la evidencia previa de asociaciones clínicas (calificación de ClinVar [29], considerando valores de ClinSig 4 o 5, correspondientes a las categorías “Likely pathogenic” y “Pathogenic”, respectivamente), el efecto fenotípico previsto, el modelo de herencia para mutaciones ya reportadas en ese gen según OMIM [34] y, si fuera posible, el efecto sobre la estructura y función proteica (predicciones de algoritmos bioinformáticos, modelado de estructuras proteicas).
Además, se buscó información adicional en Infevers [35–38] para cada una de las variantes encontradas dentro de los genes incluidos en esa base de datos. Finalmente, para las variantes reportadas en pacientes con FMF, MKD, TRAPS y CAPS, también se recurrió a las bases de datos Eurofevers [39], que recientemente incluyó información sobre la correlación genotipo-fenotipo de estas entidades.
Procesados y analizados los datos, el equipo de trabajo generó para cada caso un reporte de resultados que fue presentado al profesional médico correspondiente, quién fue responsable de entregarle una copia a la familia, orientarlos en la correcta interpretación de los datos y brindarles el asesoramiento genético pertinente.
Resultados
En el marco del proyecto PAMPA se postularon un total de 239 casos, de los cuales 165 fueron aprobados para proceder con la secuenciación exómica. En concordancia con las prevalencias reportadas en Orphanet [40] para las enfermedades autoinflamatorias más frecuentes, se obtuvo un mayor número de postulaciones con sospecha de FMF (1-5/10.000), seguidos por casos con sospecha de TRAPS (Alemania: 1/1.785.000) y, posteriormente, CAPS (Francia: 1/360.000, USA: 1-2/1.000.000). Adicionalmente, se incluyeron pacientes con sospecha de enfermedades muy poco frecuentes, entre las que podemos resaltar los Síndromes de DIRA/DITRA, PAPA, Blau, Espondilo Condrodisplasia y Síndrome H (< 1/1.000.000). En la Figura 2 se puede observar el número de casos totales postulados, y aceptados de acuerdo con el diagnóstico presuntivo. Es importante remarcar también el carácter federal del proyecto, resaltando que se recibieron casos de pacientes provenientes de numerosas provincias de Argentina.
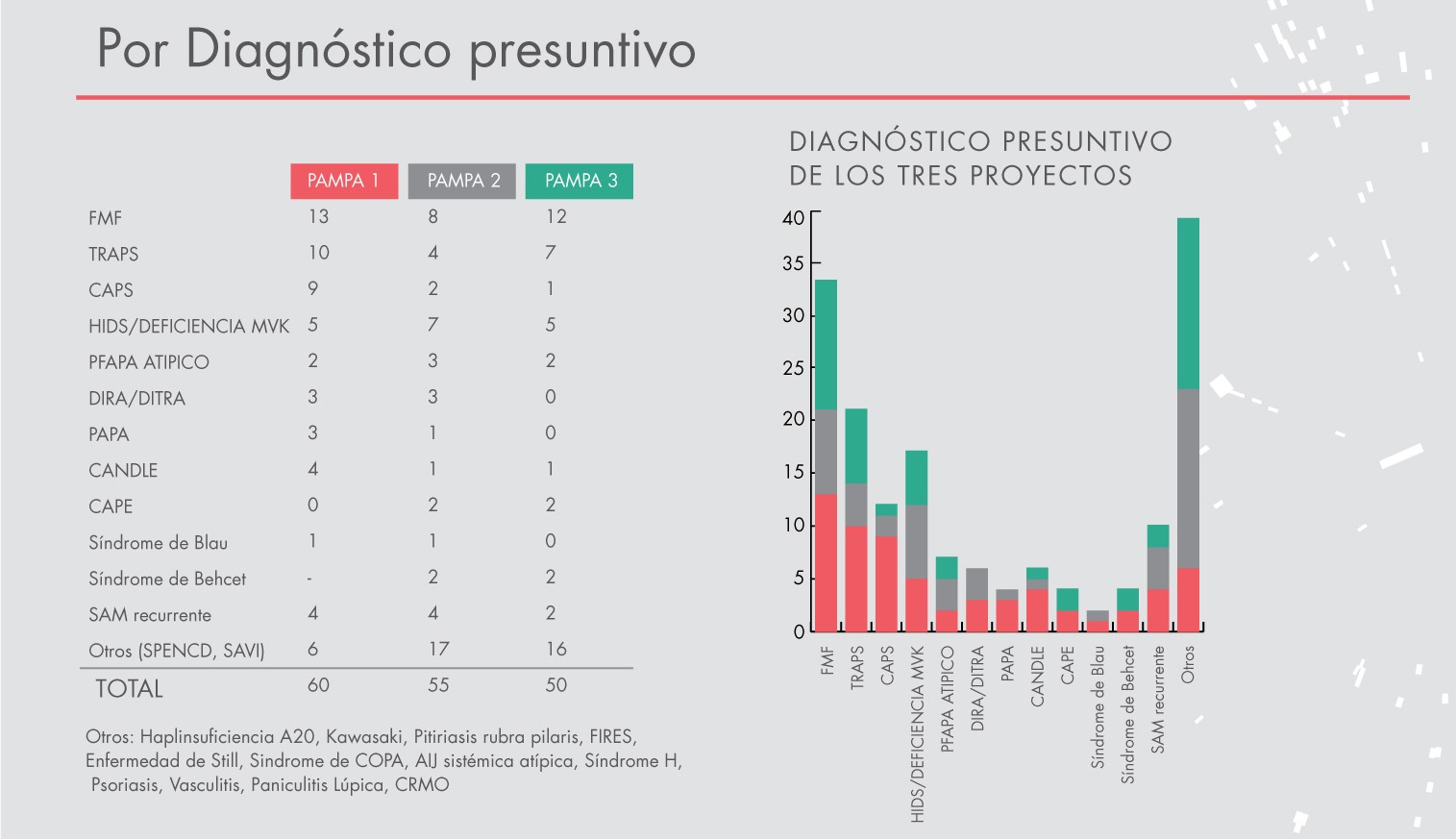
Figura 2: Número de casos aceptados agrupados en función del diagnóstico tentativo. Se observa un mayor número de postulaciones de las EAI más frecuentes, en concordancia con lo esperado. Asimismo, llama la atención el gran número de postulaciones recibidas con sospechas diagnósticas muy poco frecuentes.
Para analizar la eficiencia diagnóstica, cada caso fue clasificado en una de las siguientes categorías: “positivo”, “hipótesis de trabajo” y “negativo”, de acuerdo al grado de asociación entre el diagnóstico presuntivo (o los síntomas) del paciente y el fenotipo patológico reportado para defectos en el gen donde se encontraron la(s) variante(s) potencialmente diagnóstica(s), y teniendo en cuenta también el tipo y nivel de evidencia disponible para cada una de las variantes reportadas (Ver Tabla II).
| Resultado | Observaciones | Número de casos |
|---|---|---|
| Positivo | Se consideran como resultados ”positivos” aquellos casos en los cuales se informaron una o más variantes directamente relacionadas con el diagnóstico clínico presuntivo. | Pampa 1: 14 |
| Pampa 2: 3 | ||
| Pampa 3: 9 | ||
| Total: 26 | ||
| Hipótesis de trabajo | Se consideran como “Hipótesis de trabajo” aquellos donde se informaron alguna/s potencial/es variante/s que podría/n explicar parcialmente el fenotipo observado. | Pampa 1: 15 |
| Pampa 2: 16 | ||
| Pampa 3: 25 | ||
| Total: 56 | ||
| Negativo | Se consideran como resultados “negativos” aquellos casos donde no se encontró ninguna variante relevante para ser informada tanto en el/los gen/es directamente relacionados con la sospecha clínica, como en otros genes que podrían estar asociados con los signos y síntomas evidenciados. | Pampa 1: 31 |
| Pampa 2: 36 | ||
| Pampa 3: 16 | ||
| Total: 86 | ||
| Total | Número total de casos incorporados, secuenciados, procesados y analizados para todas las ediciones del Proyecto. | 165 |
Tabla II: Resultados de las tres Ediciones del Proyecto PAMPA. Se indica el número de casos “positivos”, “hipótesis de trabajo” y “negativos” en función del criterio mencionado.
Se consideraron como ”positivos” aquellos casos en los que se informó una (o más) variantes directamente relacionadas con la sospecha clínica y/o con el fenotipo evidenciado por el paciente, pudiendo considerarse el resultado como diagnóstico. De acuerdo a este criterio, el porcentaje de casos diagnosticados fue del 16%.
Aquellos casos en los que se informaron alguna/s potencial/es variante/s que podría/n considerarse como “hipótesis de trabajo”, y explicar parcialmente el fenotipo observado fueron agrupados en la segunda categoría, abarcando al 34% de los casos.
Finalmente, se consideraron como resultados “negativos” aquellos casos donde no se encontró ninguna variante relevante para ser informada, tanto en el/los gen/es directamente relacionados con la sospecha clínica, como en otros genes que podrían asociarse con los signos y síntomas de los pacientes estudiados (50%).
Los resultados de la Tabla III muestran 16 variantes interesantes, potencialmente asociadas a EAI, en 12 de los genes candidato estudiados (MEFV, NLRP3, MVK, NOD2, RIPK1, ACP5, SLC29A3, ISG15, TRAP1, COPA, NLRP12 y ADA2). Estos hallazgos resaltan la relevancia de este tipo de estudios sistemáticos para ampliar el conocimiento de la genética molecular subyacente a las EAI, y demuestra un gran potencial para lograr una mayor eficiencia diagnóstica futura.
| Gen | D | PR | Variantes Noveles potencialmente asociadas con EAI | |
|---|---|---|---|---|
| MEFV | 10 | 3 | 1 | NM_000243.2:c.2230G>T (p.Ala744Ser) Clasificación ACMG: VUS (PM1, PM2, PP2, BP4) |
| NLRP3 | 7 | 5 | 1 | NM_001079821.2:c.980G>A (p.Arg327Gln) Clasificación ACMG: VUS (PM2, BP4) |
| NOD2 | 5 | - | 1 | NM_022162.2:c.1558C>T (p.His520Tyr) Clasificación ACMG: Probablemente patogénica (PS2, PM1, PM2, PP3) |
| MVK | 3 | 4 | 1 | NM_000431.4:c.151C>T (p.Leu51Phe) Clasificación ACMG: VUS (PM2, PP2, PP3) |
| SLC29A3 | 2 | - | 2 | NM_018344.5:c.728T>G (p.Leu243Arg) Clasificación ACMG: VUS (PM2, PM1, PP3) NM_018344.5:c.1077_1084delCCTATGTG (p.Asp359fs) Clasificación ACMG: Patogénica (PVS1, PM2, PP3) |
| ACP5 | 2 | 1 | 1 | NM_001111036.3:c.632T>C (p.Ile211Thr) Clasificación ACMG: Probablemente Patogénica (PM1, PM2, PP2, PP3) |
| ISG15 | 2 | - | 2 | NM_005101.4:c.285del(p.Tyr96ThrfsTer5) Clasificación ACMG: Patogénica (PVS1, PS3, PM2, PM3) NM_005101.4:c.299_312del(p.Leu100Argfs) Clasificación ACMG: Patogénica (PVS1, PS3, PM2, PM3, PP3) |
| RIPK1 | 1 | 1 | 1 | NM_003804.6:c.1814T>G(p.Leu605Arg) Clasificación ACMG: Probablemente Patogénica (PM1, PM2, PP2, PP3) |
| COPA | 1 | - | 1 | NM_001098398.2:c.718T>G (p.Trp240Gly) Clasificación ACMG: VUS (PM5, PM2, PP3, BP1) |
| NLRP12 | 4 | - | 2 | NM_001277126.2:c.1463_1471del (p.Gly488_Asp490del) Clasificación ACMG: VUS (PM2, PM4, PP3) NM_001277126.1:c.3154C>T(p.Arg1052Ter) Clasificación ACMG: Patogénica (PVS1, PM2, PP3) |
| TRAP1 | 4 | - | 2 | NM_016292.2:c.319T>G (p.Ser107Ala) Clasificación ACMG: VUS (PM2, PP3, BP1) NM_016292.2:c.1711G>A (p.Ala571Thr) Clasificación ACMG: VUS/Probablemente benigna (PM2, BP1,BP4) |
| ADA2 (CECR1) | 4 | 2 | 1 | NM_001282225.2:c.145C>T(p.Arg49Trp) Clasificación ACMG: Probablemente Patogénica (PM5, PM1, PM2, PP2, PP3) |
| AP1S3 | 1 | 1 | - | |
| RBCK1 | 1 | - | - | |
| TNFAIP3 | 2 | - | - | |
| TNFRSF1A | 2 | - | - | |
| PSTPIP1 | 2 | - | - | |
| TNFRSF11A | 1 | - | - | |
Tabla III: Variantes detectadas en genes seleccionados.Se distinguen el número de variantes previamente reportadas como P/LP en la literatura/bases de datos de aquellas “noveles” que presentaron evidencia de asociación con Enfermedades Autoinflamatorias. D: Número de variantes totales detectadas. PR: Número de variantes previamente reportadas en la literatura como Patogénicas o Probablemente patogénicas.
De los 13 casos mencionados en la Tabla III, se seleccionaron 3 que resultaron de particular interés por las características de la relación genotipo-fenotipo. La descripción clínica detallada y la relevancia de los hallazgos moleculares reportados se encuentran incluidos en los BOX 1-3.
BOX 1
Un ejemplo de diagnóstico exitoso y certero lo comprende el caso de una paciente en edad pediátrica de sexo femenino, postulado por médicos genetistas, pediatras e inmunólogos de dos Hospitales de CABA, con un diagnóstico clínico presuntivo de enfermedad autoinflamatoria.
La paciente presentó fiebre aguda y autolimitada, pioderma gangrenoso, úlceras recidivantes en regiones de pliegues, sibilancias y neumonías recurrentes, alteración general del crecimiento, adenitis, leucocitosis episódica y vasculitis leucocitoclástica con cambios hipóxicos/isquémicos secundarios y ulceración epidérmica en biopsia de lesión cutánea.
Se encontraron en la paciente dos variantes frameshift en heterocigosis compuesta en el gen ISG15 (OMIM: 147571), que codifica para una proteína similar a la ubiquitina (denominada ubiquitina como modificador ISG15). Mutaciones en homocigosis o heterocigosis compuesta en este gen están asociadas con Inmunodeficiencia 38 (OMIM: 616126), una condición de herencia autosómica recesiva, que predispone a una enfermedad clínica grave por infección con micobacterias. Los individuos afectados presentan calcificaciones intracraneales, úlceras necróticas de la piel y linfadenopatías fistulizantes luego de la vacunación con BCG y poseen defectos en la inmunidad intrínseca e innata [52,53].
La primera variante c.285del (NM_005101.4), también conocida como c.284del, corresponde a una deleción de un solo nucleótido que produce un cambio de marco de lectura del exón 2 (p.Tyr96Thrfs*5) y la consecuente ganancia de un codón stop prematuro 5 residuos río abajo de la variante. La segunda variante, c.299_312del (NM_005101.4), también conocida como c.297_313del, corresponde a una deleción de 14 nucleótidos, que modifica el tamaño de la proteína resultante y produce también el desplazamiento del marco de lectura río abajo del residuo Leu100. Ambas variantes se localizaron dentro del dominio Ubiquitina-like 2, presentaron muy baja frecuencia poblacional (ausente en la base de datos de GnomAD) y no se encontraron previamente reportadas en bases de datos ni publicaciones científicas. Ambas variantes de frameshifts interrumpen claramente el segundo dominio ubiquitina, provocando la pérdida de la región C-terminal, que contiene una secuencia de aminoácidos muy conservada Leu Arg Leu Arg Gly Gly (LRLRGG), correspondiente al sitio de ISGilación y desencadenando así la pérdida de función de la proteína entera.
Según los criterios del ACMG, ambas variantes se clasificaron como patogénicas.
Este hallazgo permitió confirmar el diagnóstico molecular de la paciente, quien presentó deficiencia completa de ISG15 autosómica recesiva, seguida de lesiones cutáneas ulcerativas y enfermedad pulmonar.
El fenotipo clínico de esta paciente es único dada la presencia de manifestaciones pulmonares recurrentes y la ausencia de infecciones micobacterianas, resultando así un fenotipo distinto al descrito previamente en pacientes con variantes bialélicas de pérdida de función (LOF) en ISG15. Este caso destaca el papel de ISG15 como factor inmunomodulador cuyas variantes de LOF dan como resultado presentaciones clínicas heterogéneas. Este hallazgo motivó la incorporación de este gen al panel de autoinflamatorias.
BOX 2
Paciente pediátrico de sexo femenino, sin historia familiar de relevancia, aportado por una médica reumatóloga de un Hospital de Niños de Santa Fe. Entre los síntomas principales presentó artritis exuberante de carpos y tobillos (bilateral) con tenosinovitis marcada, dolor articular, rash maculopapular generalizado, fijo. Sarcoidosis confirmada por biopsia cutánea, BAAR negativo, PAS negativo para elementos micóticos. Se propone como diagnóstico tentativo al Síndrome de Blau.
Se detectó la variante NM_022162.2:c.1558C>T(p.His520Tyr) en heterocigosis en el gen NOD2 (OMIM:605956), que produce un cambio de nucleótido único en el exón 4, generando la sustitución del residuo Histidina 520 por Tirosina en la proteína resultante. La misma posee muy baja frecuencia poblacional (ausencia de homocigotas en GnomAD), cuenta con predicción “disease causing” por varios algoritmos bioinformáticos y se encuentra reportada en Infevers como probablemente patogénica, habiéndose detectado en un paciente sintomático con diagnóstico de Síndrome de Blau (OMIM: 186580), un trastorno de herencia autosómica dominante que se caracteriza por incluir, entre los síntomas principales, artritis granulomatosa, uveítis y rash [54]. El síndrome de Blau se asocia, usualmente, a polimorfismos de nucleótido único (SNP) de ganancia de función en el dominio NACHT del receptor inmune innato NOD2. NOD2 es un miembro de la familia de receptores de reconocimiento de patrones NLR citosólicos y constituye una proteína activada por el componente dipeptido murmil [55]. Una vez activa, interactúa con el receptor adaptador RIP2, para iniciar vías de señalización proinflamatorias que involucran el factor nuclear kappa B (NF-κB) y quinasas de estrés. De esta manera, desempeña un papel importante en la respuesta a la infección bacteriana, incluida la activación de la autofagia [55,56].
Adicionalmente, se analizó la segregación de la variante en el trío (paciente y ambos padres), demostrando la presencia de la misma en heterocigosis únicamente en la paciente. Este hallazgo aportó la evidencia necesaria que permitió reclasificar esta variante de tipo VUS a probablemente patogénica, de acuerdo a las recomendaciones y guías del ACMG. Este caso representa un ejemplo interesante en el cual la sospecha diagnóstica del profesional a cargo se termina confirmando mediante la detección de una variante probablemente patogénica a partir de la secuenciación del exoma de la paciente, en conjunto con el análisis de la segregación familiar de la misma por Sanger.
BOX 3
Paciente pediátrico de sexo femenino, sin historia familiar de relevancia, aportado por un médico inmunólogo de un Hospital de niños de Rosario. Entre los síntomas principales presentó fiebre episódica, dolor óseo, dismorfias faciales y osteoarticulares, rash, lipodistrofia, alteración general del crecimiento, esplenomegalia, reactantes de fase aguda elevados y persistentes. Como diagnóstico clínico tentativo se contempló CANDLE y, como alternativo, Síndrome H.
Se detectaron dos variantes en heterocigosis en el gen SLC29A3 (OMIM: 612373), que codifica para un transportador que regula el influjo y eflujo de nucleósidos, localizado en las membranas de lisosomas, mitocondrias y otras organelas intracelulares. Mutaciones en homocigosis o heterocigosis compuesta en este gen están asociadas con el Síndrome de histiocitosis con linfadenopatía (OMIM: 602782), un grupo de condiciones que incluye al Síndrome H y produce signos y síntomas multisistémicos compartidos. Una característica común es la histiocitosis, crecimiento y acumulación de histiocitos en los tejidos, especialmente en los ganglios linfáticos del cuello, causando inflamación (linfadenopatía). Otros síntomas pueden incluir parches cutáneos hiperpigmentados con hipertricosis, diabetes, anomalías cardíacas, hipogonadismo, hepatoesplenomegalia, bajo peso y estatura y pérdida de la audición [57]. Variantes patogénicas en el gen SLC29A3 que reducen o eliminan la actividad de la proteína codificada, conducen a la acumulación de nucleósidos en los lisosomas y otras estructuras celulares, resultando perjudicial para las mismas y afectando la producción de energía celular.
La primera variante NM_018344.5:c.728T>G(p.Leu243Arg) produce un cambio de nucleótido único en la región codificante del exón 5, generando la sustitución del residuo Leucina 243 por Arginina (un cambio significativo desde una perspectiva bioquímica), dentro del dominio transportador de nucleósidos [58]. La misma posee muy baja frecuencia poblacional, cuenta con predicción patogénica por numerosos algoritmos bioinformáticos y no se encuentra previamente descrita en bases de datos ni publicaciones científicas.
La segunda variante, NM_018344.5:c.1077_1084delCCTATGTG(p.Asp359fs), corresponde a una deleción de 8 nucleótidos, que produce el desplazamiento del marco de lectura de la región codificante del exón 6 y la ganancia de un codón stop prematuro. La misma posee muy baja frecuencia poblacional y no se encuentra reportada en bases de datos clínicas ni publicaciones científicas.
La primera variante se clasificó como de significado incierto (por falta de evidencia adicional y no por poseer propiedades que se asocian con una variante benigna) y la segunda, como probablemente patogénica.
La enfermedad autoinflamatoria causada por la presencia de ambas variantes patogénicas en SLC29A3 género en la paciente un estado de inflamación crónica con compromiso multisistémico progresivo y severo:
Hematológico: Anemia,
Dermatológico: hipertricosis, hiperpigmentación, cambios esclerodermiformes,
Oftalmológico: opacidades corneales.
Óseo: osteopenia, camptodactilia, contracturas,
Endocrinológico: hipocrecimiento severo, hasta el momento la niña sólo hizo hiperglicemias pero se debe monitorizar por su riesgo aumentado de diabetes mellitus,
Ginecológico: hipogonadismo hipogonadotrófico,
Cardíaco: taquiarritmia ventricular mejorada pero aún con riesgo aumentado de muerte súbita de origen cardíaco y necesidad de controles permanentes con cardiología y arritmología,
Dado que uno de los importantes efectos de las mutaciones en SLC29A3 es aumentar la expresión de IL-6, los inhibidores de IL-6 (por ejemplo, Tocilizumab) han demostrado ser beneficiosos en estos pacientes, por tal motivo inició tratamiento con Tocilizumab 8 mg/Kg/dosis cada 15 días según recomendaciones para Síndrome H.
En este caso, la validación de la segregación familiar de las variantes no pudo llevarse a cabo, ya que ninguno de los progenitores se encontró disponible para el análisis. Sin embargo, los hallazgos reportados aportaron información de relevancia consistente con la sospecha clínica alternativa, posibilitando el arribo a un diagnóstico molecular preciso y ampliando el espectro fenotípico del poco frecuente Síndrome H.
Desde una perspectiva molecular es de destacar que la mayoría de las variantes corresponden a variantes missense (12, siendo las restantes 3 frameshift y 1 nonsense) que son las más difíciles de interpretar y de asignar criterios de patogenicidad. Para profundizar este análisis, en la Figura 3 se muestra un score de patogenicidad que utiliza información de las distintas bases de datos como GnomAD, Clinvar y el análisis de la conservación a lo largo de las proteínas para los genes NLRP3, ISG15, NOD2 y MEFV. Con esta información se estableció un score que varía entre 0-1, el cual permite determinar zonas más o menos propensas a poseer variantes patogénicas o benignas. Como se puede observar para los 4 genes, todos presentan regiones donde las variantes tienden a ser patogénicas y regiones donde las mismas tienden a ser benignas. Interesantemente, las regiones benignas suelen ubicarse en los extremos, y específicamente en zonas con ausencia de dominios. La utilización de este análisis en relación a su contribución para la clasificación de variantes se hace evidente al analizar las variantes previamente reportadas (PR) y las potencialmente noveles encontradas en estos 4 genes en el presente trabajo. Para NLRP3, las variantes se ubican en zonas neutras (amarillas) o con tendencia a ser patogénicas (Naranja/Rojo), encontrando la variante p.Arg327Gln en una zona naranja, lo que sostiene su valor diagnóstico. Lo mismo ocurre con las variantes noveles de ISG15, NOD2 y MEFV, que se localizan en zonas con alta probabilidad de patogenicidad.
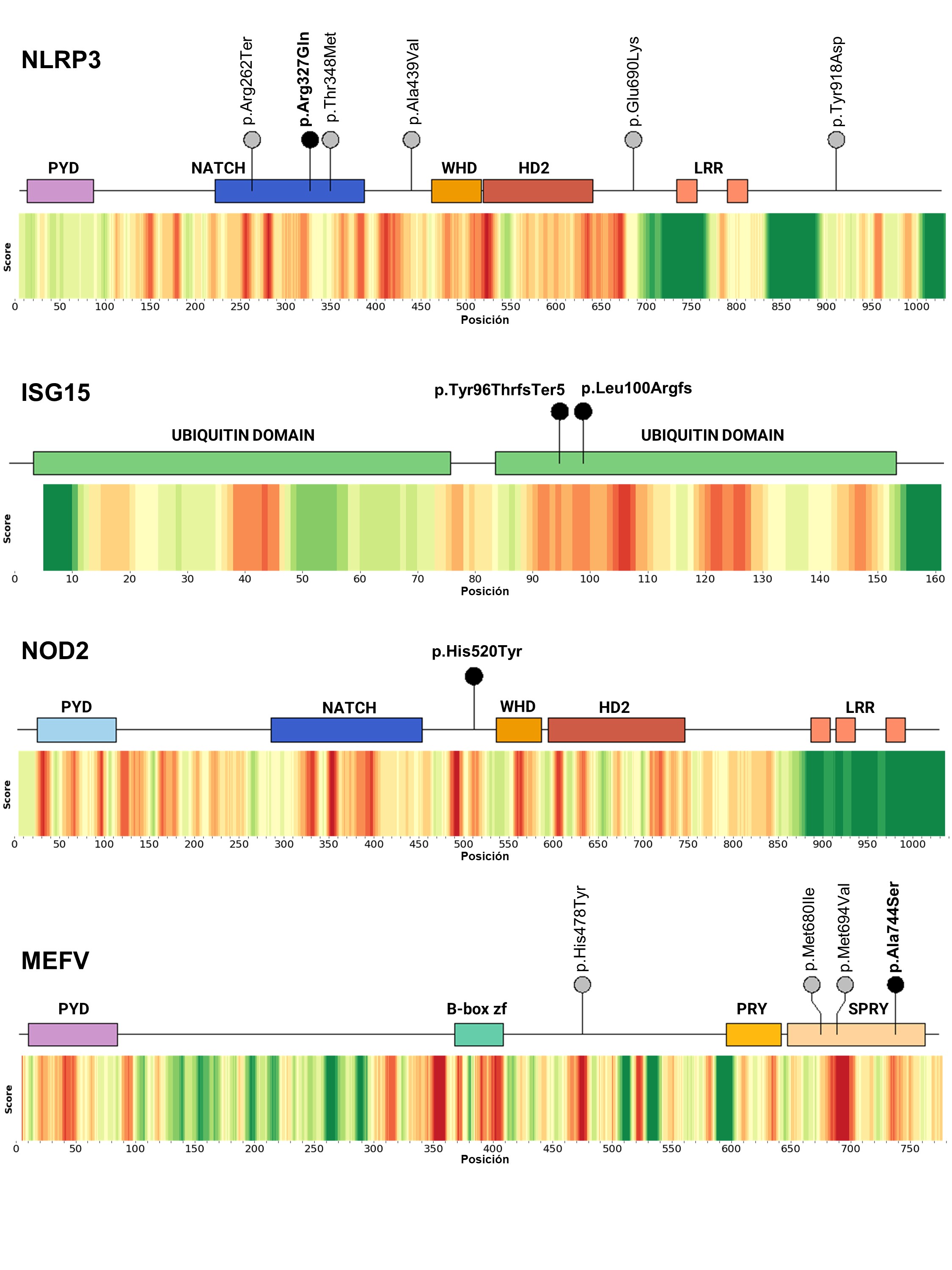
Figura 3: Representación gráfica del score de patogenicidad a lo largo de la secuencia codificante de los genes NLRP3, ISG15, NOD2 y MEFV. Naranja/Rojo indica mayor probabilidad de variante patogénica, Verde indica mayor probabilidad de variante benigna. Para cada uno de los genes se indican las variantes noveles (círculos negros) y previamente reportadas (círculos grises) encontradas en los pacientes.
Discusión
Los resultados muestran que la tasa de éxito -casos donde se arribó a un diagnóstico molecular– se estimó entre un 15-35% [42] lo que es consistente con estudios realizados a nivel internacional [43,44]. Varios de los casos, además han permitido encontrar variantes nóveles que representan el punto de partida para el estudio de los fenómenos moleculares subyacentes al desarrollo patológico, algunos de los cuales han dado lugar a publicaciones de primer nivel en el marco de colaboraciones internacionales [45,46].
Si bien los resultados de los Proyectos PAMPA no incluyeron una cantidad de casos suficiente como para realizar afirmaciones estadísticamente significativas sobre la capacidad de la secuenciación exómica para diagnosticar de manera precisa las causas moleculares de las EAI, permitieron avanzar en el descubrimiento de nuevos genes y/o la detección de nuevas variantes potencialmente patogénicas en genes con conocida asociación con estas enfermedades. Adicionalmente, se pusieron a prueba las capacidades locales con sus virtudes y dificultades, para la implementación de estas tecnologías en contextos de investigación científica e innovación clínica, y compararlos con experiencias similares realizadas en países centrales. Los resultados muestran que tanto los procesos, como la eficiencia y calidad diagnóstica son equivalentes a aquellos observados a nivel internacional.
Por otro lado, el programa posibilitó el hallazgo de algunas variantes noveles, que además de su potencial diagnóstico, constituyeron interesantes hipótesis de trabajo. Mediante la realización de estudios funcionales posteriores -como se ha realizado para las variantes detectadas en ISG15- se buscó profundizar nuestra comprensión de los mecanismos asociados a la etiopatologìa molecular de las EAI y al funcionamiento del sistema inmune a nivel molecular-celular. Además, el análisis de las variantes noveles de tipo missense (tanto las reportadas como las descartadas), en el contexto de aquellas conocidas y asignadas como Patogénicas o Benignas, permitió obtener una mayor comprensión del potencial efecto de las mismas, contribuyendo a evaluar la gran cantidad de variantes de significado incierto detectadas en estos genes.
Finalmente, y desde una perspectiva sociológica, la experiencia de estos proyectos permitió darle peso y reforzar la siguiente filosofía de trabajo interdisciplinario: si bien la división de tareas es un proceso lógico, práctico y adecuado para muchos trabajos, tales como el relevamiento de la historia clínica por parte del médico, la toma y procesamiento de la muestra por el bioquímico de laboratorio, el procesamiento de datos por parte del bioinformático, la priorización de variantes por parte de bioquímicos/biólogos moleculares/médicos genetistas y finalmente la interpretación de las mismas y devolución al paciente por parte del médico; la eficiencia y las chances de éxito se maximizan cuando todos los profesionales trabajan en conjunto. Particularmente, esto se evidencia tanto en la etapa de postulación y selección de pacientes como en la priorización e interpretación de las variantes, la cual requiere integrar conocimientos asociados al experimento de NGS (cobertura, profundidad, calidad de la variante, entre otras), al impacto a nivel molecular de las mismas y, por supuesto, a su potencial relación con la clínica. A lo largo de estos 3 años de experiencia, destacamos principalmente el carácter federal del programa, que brindó la posibilidad a pacientes de diversos puntos del país de incorporarse a un proyecto de medicina de precisión financiado por Novartis. Adicionalmente, queremos resaltar que la probabilidad de éxito se vio incrementada en aquellos casos en los que se evidenció la identificación de un diagnóstico presuntivo preciso, y el profundo conocimiento de los genes asociados a enfermedades autoinflamatorias por parte del equipo médico postulante.
Referencias:
1. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. (2018) The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol 38: 129–143. DOI: 10.1007/s10875-017-0465-8
2. Ciccarelli F, Martinis M, Ginaldi L (2013) An Update on Autoinflammatory Diseases. Current Medicinal Chemistry 21: 261–269. DOI:10.2174/09298673113206660303
3. Kastner DL, Aksentijevich I, Goldbach-Mansky R (2010) Autoinflammatory disease reloaded: a clinical perspective. Cell 140: 784–790. DOI:10.1016/j.cell.2010.03.002
4. Demir S, Sag E, Dedeoglu F, Ozen S (2018) Vasculitis in Systemic Autoinflammatory Diseases. Front Pediatr 6: 377. DOI: 10.3389/fped.2018.00377
5. Davidson S, Steiner A, Harapas CR, Masters SL (2018) An Update on Autoinflammatory Diseases: Interferonopathies. Curr Rheumatol Rep 20: 38. DOI: 10.1007/s11926-018-0748-y
6. Harapas CR, Steiner A, Davidson S, Masters SL (2018) An Update on Autoinflammatory Diseases: Inflammasomopathies. Curr Rheumatol Rep 20: 40. DOI: 10.1007/s11926-018-0750-4
7. Steiner A, Harapas CR, Masters SL, Davidson S (2018) An Update on Autoinflammatory Diseases: Relopathies. Curr Rheumatol Rep 20: 39. DOI: 10.1007/s11926-018-0749-x
8. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL (2017) Corrigendum: The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 18: 1271. DOI: 10.1038/ni.3777
9. https://sademi.com/wp-content/uploads/2016/01/Cuadernos-Autoinmunidad-A%C3%B1o-13-num-2.pdf, accesado 17/09/2021
10. Al-Mayouf S, Alsonbul A (2015) Monogenic and multifactorial autoinflammatory diseases: Clinical and laboratory characterization in a pediatric Saudi population. Pediatric Rheumatology 13(Suppl 1): P160. DOI: 10.1186/1546-0096-13-s1-p160
11. Vanoni F, Theodoropoulou K, Hofer M (2016) PFAPA syndrome: a review on treatment and outcome. Pediatr Rheumatol Online J 14: 38. DOI: 10.1186/s12969-016-0101-9
12. Labrousse M, Kevorkian-Verguet C, Boursier G, Rowczenio D, Maurier F, Lazaro E, et al. (2018) Mosaicism in autoinflammatory diseases: Cryopyrin-associated periodic syndromes (CAPS) and beyond. A systematic review. Crit Rev Clin Lab Sci 55: 432–442. DOI: 10.1080/10408363.2018.1488805
13. Hoffman HM, Broderick L (2017) Editorial: It Just Takes One: Somatic Mosaicism in Autoinflammatory Disease. Arthritis Rheumatol 69: 253–256. DOI: 10.1002/art.39961
14. Ryan JG, Goldbach-Mansky R (2008) The spectrum of autoinflammatory diseases: recent bench to bedside observations. Curr Opin Rheumatol 20: 66–75. DOI: 10.1097/BOR.0b013e3282f1bf4b
15. Jesus AA, Goldbach-Mansky R (2014) IL-1 Blockade in Autoinflammatory Syndromes. Annual Review of Medicine 65: 223–244. DOI: 10.1146/annurev-med-061512-150641
16. Caorsi R, Federici S, Gattorno M (2012) Biologic drugs in autoinflammatory syndromes. Autoimmun Rev 12: 81–86. DOI: 10.1016/j.autrev.2012.07.027
17. Reinhardt RL, Liang H-E, Bao K, Price AE, Mohrs M, Kelly BL, et al. (2015) A novel model for IFN-γ-mediated autoinflammatory syndromes. J Immunol 194: 2358–2368. DOI: 10.4049/jimmunol.1401992
18. Efthimiou P (2019) Auto-Inflammatory Syndromes: Pathophysiology, Diagnosis, and Management. Springer.
19. Federici S, Gattorno M (2014) A practical approach to the diagnosis of autoinflammatory diseases in childhood. Best Practice & Research Clinical Rheumatology 28: 263–276. DOI:10.1016/j.berh.2014.05.005
20. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. (2019) Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis 78: 1025–1032. DOI: 10.1136/annrheumdis-2019-215048
21. Holzer FS, Mastroleo ID (2018) Ethical Aspects of Precision Medicine: An Introduction to the Ethics and Concept of Clinical Innovation. Precision Medicine 1–19. DOI:10.1016/b978-0-12-805364-5.00001-9
22. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. DOI: 10.1093/bioinformatics/btp324
23. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. DOI: 10.1101/gr.107524.110
24. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. DOI: 10.1038/ng.806
25. Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy‐Moonshine A, et al. (2013) From FastQ Data to High‐Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Current Protocols in Bioinformatics 43: 1-33. DOI:10.1002/0471250953.bi1110s43
26. Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6: 80–92. DOI: 10.4161/fly.19695
27. Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38: e164. DOI: 10.1093/nar/gkq603
28. Lek M, Karczewski KJ, Minikel EV, Samocha KE, et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285-291. DOI:http://dx.doi.org/10.3410/f.726646180.793565293 10.1038/nature19057
29. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44: D862–8. DOI: 10.1093/nar/gkv1222
30. Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 7: Unit7.20. DOI: 10.1002/0471142905.hg0720s76
31. Donev R (2022) Advances in Protein Chemistry and Structural Biology. Inmunotherapeutics. Academic Press 129: 1-477
32. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7: 575–576. DOI: 10.1038/nmeth0810-575
33. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. DOI: 10.1038/gim.2015.30
34. https://omim.org/, accesado 20/09/2021
35. Van Gijn ME, Ceccherini I, Shinar Y, Carbo EC, Slofstra M, Arostegui JI, et al. (2018) New workflow for classification of genetic variants’ pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). Journal of Medical Genetics 55: 530–537. DOI:10.1136/jmedgenet-2017-105216
36. Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, et al. (2008) The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat 29: 803–808. DOI: 10.1002/humu.20720
37. Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, et al. (2004) Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Muta 24: 194–198. DOI: 10.1002/humu.20080
38. Sarrauste de Menthière C, Terrière S, Pugnère D, Ruiz M, Demaille J, Touitou I (2003) INFEVERS: the Registry for FMF and hereditary inflammatory disorders mutations. Nucleic Acids Res 31: 282–285. DOI: 10.1093/nar/gkg031
39. https://www.printo.it/eurofever/registry, accesado 08/08/2021
40. http://www.orpha.net, accesado 21/08/2017
41. den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. (2016) HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat 37: 564–569. DOI: 10.1002/humu.22981
42. Zhang X (2014) Exome sequencing greatly expedites the progressive research of Mendelian diseases. Front Med. 8: 42–57. DOI: 10.1007/s11684-014-0303-9
43. Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, et al. (2016) Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet 89: 275–284. DOI: 10.1111/cge.12654
44. Kosukcu C, Taskiran EZ, Batu ED, Sag E, Bilginer Y, Alikasifoglu M, et al. (2021) Whole exome sequencing in unclassified autoinflammatory diseases: more monogenic diseases in the pipeline?. Rheumatology 60: 607–616. DOI: 10.1093/rheumatology/keaa165
45. Buda G, Valdez RM, Biagioli G, Olivieri FA, Affranchino N, Bouso C, et al. (2020) Inflammatory cutaneous lesions and pulmonary manifestations in a new patient with autosomal recessive ISG15 deficiency case report. Allergy, Asthma & Clinical Immunology 16:77. DOI: 10.1186/s13223-020-00473-7
46. Martin-Fernandez M, Bravo García-Morato M, Gruber C, Murias Loza S, Malik MNH, Alsohime F, et al. (2020) Systemic Type I IFN Inflammation in Human ISG15 Deficiency Leads to Necrotizing Skin Lesions. Cell Rep 31: 107633. DOI: 10.1016/j.celrep.2020.107633
47. Shinar Y, Giat E, Cohen R, Livneh A (2015) Assessment of the pathogenicity of the p.K695R and p.A744S Mediterranean fever gene variants. Pediatric Rheumatology 13 (Suppl 1): P122. DOI:10.1186/1546-0096-13-s1-p122
48. Fokkema IFAC, Fokkema IFA, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, et al. (2011) LOVD v.2.0: the next generation in gene variant databases. Human Mutation. 32: 557–563. DOI:10.1002/humu.21438
49. Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, et al. (2020) Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577: 103–108. DOI: 10.1038/s41586-019-1828-5
50. Standing ASI, Hong Y, Paisan-Ruiz C, Omoyinmi E, Medlar A, Stanescu H, et al. (2020) TRAP1 chaperone protein mutations and autoinflammation. Life Science Alliance 3: e201900376. DOI:10.26508/lsa.201900376
51. Burillo-Sanz S, Montes-Cano M-A, García-Lozano J-R, Ortiz-Fernández L, Ortego-Centeno N, García-Hernández F-J, et al. (2017) Mutational profile of rare variants in inflammasome-related genes in Behçet disease: A Next Generation Sequencing approach. Sci Rep. 7: 8453. DOI: 10.1038/s41598-017-09164-7
52. Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. (2012) Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337: 1684–1688. DOI: 10.1126/science.1224026
53. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. (2015) Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517: 89–93. DOI: 10.1038/nature13801
54. Parkhouse R, Boyle JP, Monie TP. (2014) Blau syndrome polymorphisms in NOD2 identify nucleotide hydrolysis and helical domain 1 as signalling regulators. FEBS Lett. 588: 3382–3389. DOI: 10.1016/j.febslet.2014.07.029
55. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278: 8869–8872. DOI: 10.1074/jbc.C200651200
56. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE (2014) NOD proteins: regulators of inflammation in health and disease. Nature Reviews Immunology 14: 9–23. DOI:10.1038/nri3565
57. Molho-Pessach V, Ramot Y, Camille F, Doviner V, Babay S, Luis SJ, et al. (2014) H syndrome: the first 79 patients. J Am Acad Dermatol 70: 80–88. DOI: 10.1016/j.jaad.2013.09.019
58. Limviphuvadh V, Tan CS, Konishi F, Jenjaroenpun P, Xiang JS, Kremenska Y, et al. (2018) Discovering novel SNPs that are correlated with patient outcome in a Singaporean cancer patient cohort treated with gemcitabine-based chemotherapy. BMC Cancer 18: 1–16. DOI: 10.1186/s12885-018-4471-x
|
Revista QuímicaViva Número 2, año 21, Agosto 2022 quimicaviva@qb.fcen.uba.ar |