El genoma del coronavirus
Agustín M. Pardo2,3, Claudio Schuster2,3, María Mercedes Palomino2,3, Adrián Turjanski2,3, Darío A. Fernández Do Porto1,2,#
1Instituto de Cálculo, 2Departamento de Química Biológica, 3Instituto de Química Biológica (IQUIBICEN). Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Argentina.
Resumen
En enero de 2020 se reportaría oficialmente el agente causal de una nueva y misteriosa neumonía, una variante de coronavirus: SARS-CoV-2. En pocas semanas e incluso días luego de la aparición del virus, originado en la ciudad China de Wuhan, comenzaron a manifestarse ininterrumpidamente brotes en diferentes lugares de mundo. Casi a la par que esto sucedía, fueron obtenidas las secuencias genómicas de decenas de miles de aislamientos clínicos de los cinco continentes. El hecho de que las secuencias del genoma se obtengan sincrónicamente con el desarrollo de la pandemia permite estudiar el proceso evolutivo de manera simultánea al desarrollo de la misma. Esto representa un evento sin precedentes para la historia científica de la humanidad. En este artículo exploramos la importancia de contar con esta información y cómo nos ayuda a entender el desarrollo de esta pandemia.
Palabras clave: SARS-CoV-2, genómica, filogenia.
The coronavirus genome
Summary
In January 2020, the causal agent of a mysterious new pneumonia, a variant of coronavirus (SARS-CoV-2), was officially reported. After the first appearance of the virus in Wuhan (China), different outbreaks started throughout the world. Almost simultaneously, viral genomes of thousands of clinical isolates were globally sequenced. These genome sequences are being obtained simultaneously with the pandemic allowing us to study the pandemic evolution at the same time of its development. This represents an unprecedented event for human scientific history. In this article we explore the importance of these data and how it can help us to understand the development of SARS-CoV-2 pandemic.
Keywords: SARS-CoV-2, genomics, phylogeny
En diciembre de 2019 un grupo de casos relacionados a una misteriosa neumonía fueron reportados en las cercanías de un mercado de mariscos en Wuhan, China (recientemente reabierto). En enero de 2020 se informó oficialmente el agente causal de estas infecciones respiratorias, una nueva variante de coronavirus: el coronavirus 2 del síndrome respiratorio agudo grave, del inglés, severe acute respiratory syndrome coronavirus (SARS-CoV-2).
El primer reporte de coronavirus como agente patógeno humano se remonta a mediados de los años 60, cuando Tyrrell y Bynoe lograron aislarlo por primera vez a partir de muestras del tracto respiratorio de personas con síntomas de resfriado común.
Estos virus de la Familia Coronaviridae del Orden Nidovirales, deben su nombre al hecho de tener una forma esférica de la que sobresalen unas espículas que les dan la apariencia de una corona. Esta “corona” encierra las instrucciones genéticas para hacer millones de copias de sí misma, información codificada en 30.000 “letras” de RNA (A, C, G y U) [1]. Esta información es la que conocemos como genoma. El virus una vez que infecta la célula hospedadora utiliza la maquinaria de la célula infectada “obligándola” a “fabricar” los distintos tipos de proteínas virales.
COVID-19, enfermedad causada por SARS-CoV-2, es la tercera infección zoonótica de coronavirus conocida después del Síndrome Agudo Respiratorio Severo (SARS), causada por SARS-CoV-1, y el Síndrome Respiratorio del Medio Oriente (MERS), causada por MERS-CoV. Estos tres coronavirus pertenecen al grupo de β-coronavirus (Envuelto, RNA de sentido positivo y no segmentado) [2]. Los genomas de Coronavirus (~26–32 kilobases) constituyen los RNA genómicos más largos que se conocen a la fecha. En enero de 2020 científicos chinos obtuvieron la secuencia de “letras” (secuenciación) del RNA de SARS-CoV-2, aislado de un hombre que trabajaba en el mercado de Wuhan (identificador del NCBI database NC_045512.2). Ese primer genoma se convirtió en la referencia contra la cual se contrastan todos los genomas secuenciados de SARS-CoV-2.

Figura 1: Estructura del genoma del coronavirus de Wuhan.
Este genoma de referencia tiene una longitud de 29,9 kb. Luego de una primera región no codificante de 265 nucleótidos, dos marcos de lectura abiertos: ORF1a y ORF1ab, que corresponden a dos terceras partes del RNA viral y codifican para dos poliproteínas, pp1a y pp1ab, las cuales a través de un proceso de clivaje dan origen a por lo menos 16 proteínas no estructurales (nsp1 a nsp16). Las funciones relacionadas con la síntesis y el procesamiento del RNA residen en las proteínas nsp 7 a nsp 16. La parte restante del genoma del virus codifica para cuatro proteínas estructurales esenciales, incluida la glicoproteína de la espiga (S) (que forman las espículas de la “corona”), la proteína de envoltura pequeña (E), la proteína de la matriz (M) y la proteína de la nucleocápside (N), y también varias proteínas accesorias, que interfieren con la respuesta inmune innata del hospedador [3]. El genoma completo de coronavirus puede ser navegado dentro de la plataforma de desarrollo nacional Target Pathogen (http://target.sbg.qb.fcen.uba.ar/patho/) [4].
Mutaciones en el RNA
Una célula infectada libera millones de nuevos coronavirus, todos son copias creadas a partir del RNA genómico original. Cuando la célula infectada “copia” el genoma viral suele cometer errores que generalmente consisten en una sola “letra” equivocada. Estos errores son llamados mutaciones o variantes (cuando se producen en una sola “letra” se las denomina mutaciones de nucleótido único). A medida que los coronavirus se transmiten de persona a persona y se produce el sucesivo “copiado”, acumulan mutaciones al azar. Estas mutaciones son las que utilizan los investigadores para rastrear la propagación del virus por el mundo. Las nuevas mutaciones se acumulan en los virus a un ritmo más o menos regular, por lo que a partir de la tasa de mutación viral se pudo inferir que el origen del brote fue anterior a diciembre de 2019.
Un genoma secuenciado a continuación, proveniente de otro de los primeros pacientes de Wuhan, era idéntico al primer caso, excepto por una mutación. En la posición 186 del RNA viral se encontraba una U en lugar de una C (esta región corresponde a la posición no codificante 5’).
Esta misma mutación se encontró en una muestra recolectada casi dos meses después en la región de China denominada Guangzhou. Este hecho nos da una idea que la muestra de Guangzhou sería descendiente directa de la primera muestra de Wuhan o que tanto la muestra de Wuhan como lo de Guangzhou compartían un ancestro común. También podría ser el caso de que en ambos genomas hubiera ocurrido azarosamente la misma mutación “de novo”, un fenómeno conocido como homoplasia. Sin embargo, la acumulación de las mismas mutaciones “de novo” en dos aislamientos virales separados temporal y geográficamente es un fenómeno poco probable, por lo que dos muestras que han adquirido las mismas variantes con respecto al genoma de referencia probablemente se encuentren emparentadas. En estas suposiciones se basan los científicos para poder predecir el origen y propagación de la pandemia.
El linaje de Guangzhou continuó saltando de persona a persona. En el camino, desarrolló dos nuevas variantes: dos letras más del RNA cambiaron a U.
A diferencia de la primera mutación, las dos nuevas encontradas en los genomas de Guangzhou sí se encontraban en regiones codificantes que modificarían las proteínas del virus.
Las proteínas son cadenas lineales de aminoácidos que se pliegan en el espacio adoptando distintas conformaciones. Cada aminoácido está codificado por tres letras en el RNA (denominados codones). Sin embargo, en algunos casos, una variante en la tercera letra de un codón seguirá codificando el mismo aminoácido. Estas mutaciones llamadas “silenciosas” no producen cambios en la proteína resultante.
Las mutaciones “no silenciosas” sí cambian los aminoácidos de una proteína. De todas formas, cambiar un solo aminoácido de la proteína no siempre varía su forma o funcionamiento.
Son estas mutaciones que se van acumulando en los genomas del coronavirus lo que permite a los científicos rastrear la propagación del virus alrededor del mundo.
Desde que la primera secuencia de genoma completo del nuevo coronavirus, SARS-CoV-2, se compartió en línea el 12 de enero, los científicos han secuenciado y puesto a disposición, a través del repositorio de GISAID (Global Initiative on Sharing All Influenza and Covid-19 Data, gisaid.org) [5], más de 45,000 genomas virales de todo el mundo. Esta gran cantidad de datos ha permitido a los investigadores rastrear el origen de los brotes de SARS-CoV-2 en sus países y determinar cuándo se produjo la transmisión comunitaria. A continuación se muestra un captura de pantalla de la plataforma de GISAID mostrando la circulación del virus a nivel mundial.
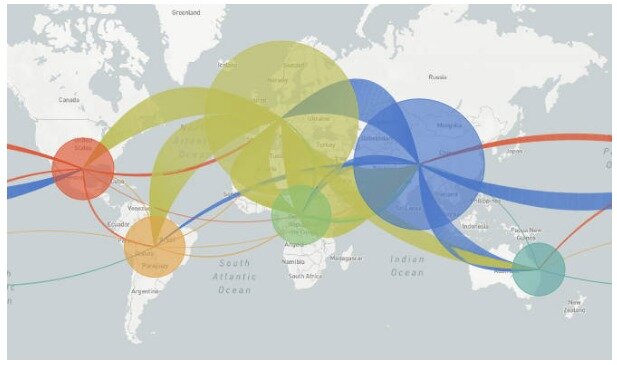
Figura 2: Circulación del coronavirus a nivel mundial. Toma de captura del repositorio de GISAID al 12 de junio de 2020.
Las primeras secuencias genómicas en Argentina
A principios del mes de abril, científicos y técnicos de la Administración Nacional de Laboratorios e Institutos de Salud (ANLIS) del Instituto Malbrán secuenciaron los primeros 3 genomas completos del SARS CoV-2 de pacientes infectados. Tiempo después, se conformó el Consorcio Argentino de Genómica de SARS-CoV-2 del cual forman parte los autores de este proyecto. En primera instancia se secuenciaron 26, pero se planea a corto plazo un muestreo en todo el país de aproximadamente 1000 genomas de SARS-CoV-2.
¿Cómo podemos ayudar a entender mejor el desarrollo de una pandemia desde la genómica?
Como comentamos anteriormente podemos estudiar la acumulación de mutaciones en los genomas de los aislamientos de pacientes separados temporal y geográficamente. Las mutaciones en el genoma viral se dan al azar y se conservan en las replicaciones sucesivas del virus. Esto nos permite realizar comparaciones de dichas variantes para decidir qué secuencias genómicas se parecen más entre sí y por lo tanto “rastrear su origen”, o mejor dicho predecir a qué regiones se parecen las secuencias que circulan en nuestro país.
Tomemos el ejemplo de 3 genomas de coronavirus provenientes de pacientes de Argentina (Identificador de base de datos GISAID (https://www.gisaid.org/): 420598, 430795, 430817) y 4 genomas de países de diferentes regiones, Estados Unidos (USA, 437513), Países Bajos (461359), Brasil (427298), y la referencia proveniente de China, Wuhan (identificador del NCBI database NC_045512.2). Al analizar esos genomas podemos encontrar variantes (respecto del genoma de referencia de Wuhan) que sólo son compartidas por ciertas muestras (Tabla 1).
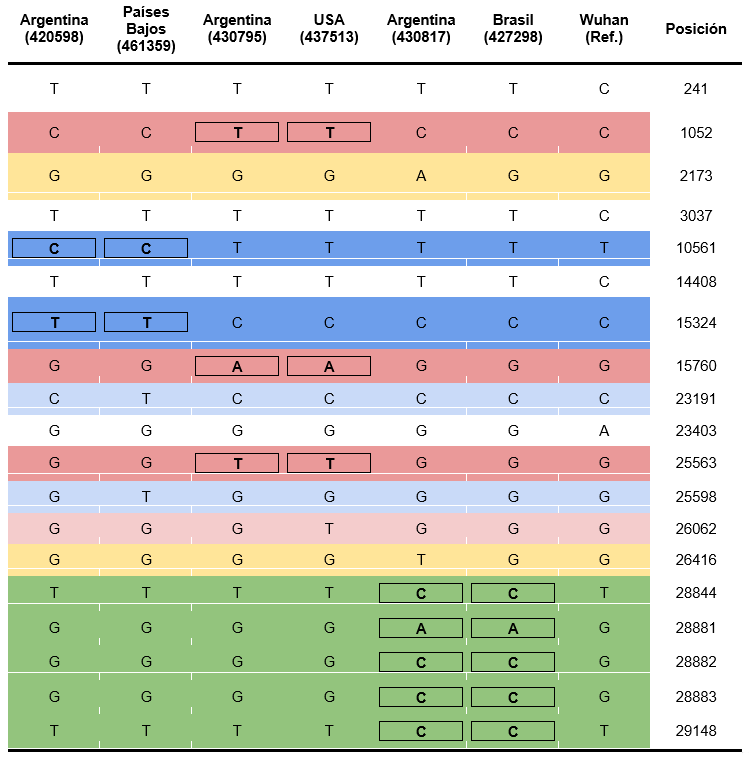
Tabla 1: Variantes presentes en tres genomas argentinos (420598, 430795 y 430817) y tres genomas filogenéticamente cercanos de tres regiones diferentes (Países Bajos, Estados Unidos y Brasil) con respecto al genoma de referencia de Wuhan. “Posición” indica la posición de la variante respecto al genoma de referencia. Referencia de colores: Blanco, variantes que comparten todas menos la referencia. Rojo claro, variante única de 437513 (USA). Rojo oscuro, variante que comparten únicamente las secuencias 437513 (Estados Unidos) y 430795 (Argentina). Azul claro, variante única de la secuencia 461359 (Países Bajos). Azul oscuro, variantes compartidas entre el genoma 430817 (Argentina) y 461359 (Países Bajos). Verde oscuro, variantes compartidas entre las secuencias 430817 (Argentina) y 427298 (Brasil). Amarillo claro, variantes únicas de 430817 (Argentina). Se resalta en negrita y con un recuadro las variantes compartidas. Toma de captura del repositorio de GISAID al 12 de junio de 2020.
En la Tabla 1 se observa que hay un vínculo genético entre las secuencias 437513 (USA) y 430795 (Argentina) dado que comparten las mismas variantes en las posiciones 1052, 15760 y 25563, que a su vez, no se encuentran presentes en las otras secuencias comparadas. La misma relación la podemos encontrar entre las secuencias 461359 (Países Bajos) y 420598 (comparten variantes en las posiciones 10561, 15324), y Brasil con 430817 (por las variantes en las posiciones 28844, 28881, 28882, 28883).
A su vez cabe destacar que las variantes presentes en las posiciones 241, 3037, 14408 y 23403 se encuentran presentes en todas las cepas estudiadas (aún siendo filogenéticamente distantes) lo que indicaría que estas mutaciones han sido adquiridas de manera temprana en la evolución viral. Por otro lado, se puede observar que no existen variantes (que no sean las cuatro adquiridas tempranamente), compartidas en los tres genomas argentinos analizados, lo que revela que las tres cepas tienen origen diferente (es decir entraron al país a través de diferentes eventos). El bajo número de variantes presentes únicamente en las cepas de nuestro territorio también nos da una idea del poco tiempo transcurrido entre la entrada del virus al país y la toma de muestra en los pacientes de los cuales se obtuvieron estos genomas.
Reconstruyendo la filogenia
A partir de un análisis como el antes comentado se puede inferir un árbol filogenético para representar las relaciones evolutivas entre las cepas secuenciadas. En estos árboles, cada rama representa un genoma viral. La disponibilidad de las secuencias genómicas permite agrupar aquellas secuencias que tienen variantes en común, lo que nos da una idea de qué tan similares o qué tan relacionadas están unas de otras.
A continuación, vemos una ilustración (Figura 3) que representa el árbol filogenético de las secuencias analizadas en la Tabla 1. A la izquierda se encuentra el árbol y con círculos de colores las variantes que caracterizan a cada rama del árbol, esto es, qué variantes son las que crean la bifurcación en las ramas. A la derecha figuran las representaciones de las secuencias también con variantes que se muestran como círculos de colores. En ambos casos los colores respetan el análisis realizado en la Tabla 1. Podemos ver que las secuencias que comparten las mismas mutaciones se agrupan en ramas más cercanas el árbol. Cuanto mas cercanas las ramas del árbol más parecidas son las secuencias entre sí. Por ejemplo las secuencias 420596 (Argentina) y 461359 (Países Bajos) presentan ciertas variantes que comparten entre sí (azul oscuro) lo que las hace estar más cercanas en el árbol filogenético que el resto. A su vez la secuencia 461359 (Países Bajos) posee variantes que no comparte con la Argentina (420596) ni con el resto de las secuencias (azul claro) lo que indica que esta secuencia no es la misma que la de Argentina (420596) creando una bifurcación en el árbol filogenético. Se puede observar en el árbol que en dicha bifurcación se posa el círculo celeste claro, entendiéndose que dichas variantes son responsables de la bifurcación del árbol y de que dichas secuencias no sean idénticas.
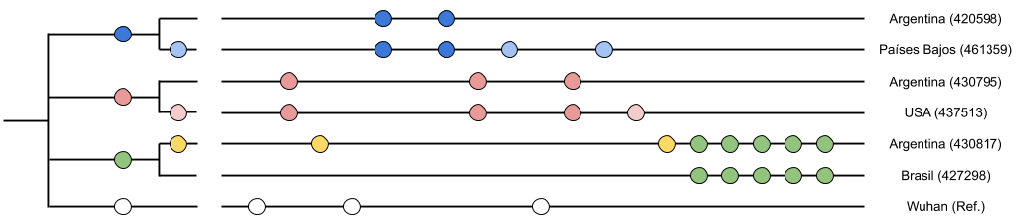
Figura 3: Árbol filogenético construido a partir de las secuencias de la Tabla 1 y sus variantes. La figura fue construida a partir de las secuencias argentinas 420598, 430795, 430817 y 4 genomas de países de diferentes regiones, Estados Unidos (USA, 437513), Países Bajos (461359), Brasil (427298), y la referencia proveniente de China, Wuhan. A la izquierda se encuentra un árbol filogenético, a la derecha una representación del genoma de las secuencias que conforman el árbol. Las variantes en cada secuencia se indican con círculos de colores tanto en el árbol como en las secuencias, respetando el análisis realizado en la Tabla 1.
En la actualidad la comunidad científica deposita los genomas secuenciados en la base de datos de GISAID. A partir de dichos genomas se realiza el análisis epidemiológico genómico en la plataforma Nextstrain (https://nextstrain.org/ncov/global) [6]. En dicha plataforma se visualiza un árbol filogenético construido a partir de 4718 genomas de SARS-CoV-2 provenientes de la base de datos GISAID.
En la Figura 4 se observa capturas de pantallas del árbol filogenético yendo desde una vista general hasta un enfoque particular de la secuencia argentina 42509. En éste último se puede observar la cercanía entre la secuencia argentina 425098 y la secuencia de Países Bajos antes mencionada.
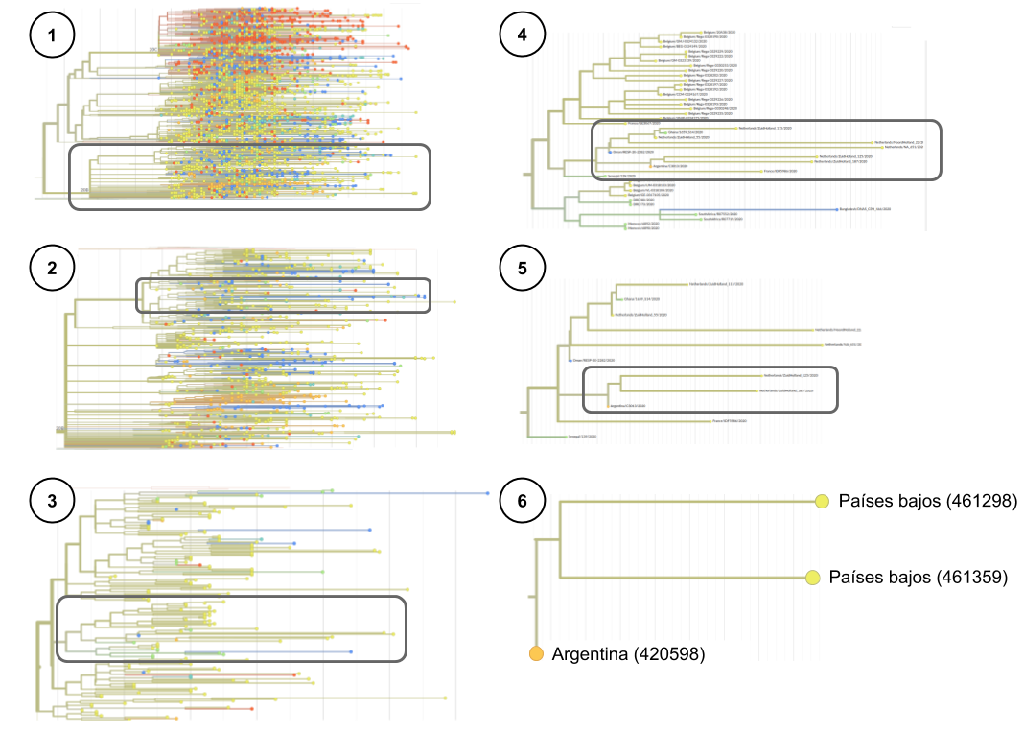
Figura 4: Capturas de pantalla tomadas recorriendo la plataforma de Nextstrain. Se observan distintos aumentos sucesivos de escala (zoom) del árbol filogenético de SARS-CoV-2, siendo 1 la base o raíz del árbol y 6 el aumento más grande (haciendo foco en la secuencia argentina 420598). Los colores de los nodos indican la procedencia de cada secuencia. A su vez el color de cada rama indica la procedencia mayoritaria de los nodos que contiene, siendo azul de Asia, turquesa de Oceanía, verde de Africa, amarillo de Europa, naranja de Sudamérica, y rojo de Norteamérica.
Por otra parte, la topología del árbol indica las relaciones filogenéticas de las ramas. Cuanto más próximas, más emparentadas. En este caso, la cercanía de la rama de Países Bajos y de Argentina indica un parentesco o relación filogenética muy cercano (Figura 4, recuadro 6). El nodo del árbol de donde se desprende tanto la secuencia de Argentina como la de Países Bajos es el ancestro común de ambas, un ancestro hipotético del cual evolucionaron ambas secuencias.
¿Qué sucede en nuestra región?
A los fines de facilitar el entendimiento de los parentescos filogenéticos de las secuencias se establecen sistemas de clasificación por linajes. En esencia, a partir de un análisis filogenético se asignan nombres a ciertas ramas. De este modo, al decir que una secuencia forma parte de un determinado linaje, es posible entender su ubicación en el árbol rápidamente y con qué otras secuencias está más emparentada.
Una manera de asignar linaje a una secuencia en particular de manera rápida es mediante el programa Pangolin (https://pangolin.cog-uk.io/). Este programa toma una secuencia a analizar, la incluye en un alineamiento de secuencias representativas de todos los linajes preestablecidos [7] y luego computa una filogenia utilizando un árbol inicial guía que es representativo de la filogenia de los linajes. Una vez obtenido un árbol definitivo, Pangolin determina el linaje de la secuencia en función de su ubicación en el árbol y del linaje al que pertenecen sus vecinos más próximos.
Al completar el análisis, Pangolin devuelve una tabla donde cada fila es una de las secuencias analizadas y las columnas corresponden al nombre del archivo donde estaba la secuencia, el nombre de la secuencia, el linaje asignado y los valores de soporte de ramas de Bootstrap [8] y SH-aLRT [9]. Estos dos últimos son estadísticos indicativos de la “credibilidad” de una rama del árbol.
Las secuencias del genoma de SARS-CoV-2 obtenidas a partir de individuos infectados de distintos países han sido clasificadas en dos linajes principales (denominados con letras A y B) y varios linajes internos (A1-A5 y sus subgrupos, o B1-B8 y sus subgrupos) de acuerdo con cambios nucleotídicos y su agrupamiento filogenético [7]. En esencia, estos linajes constituyen grupos monofiléticos definidos a partir de una filogenia realizada con una serie de secuencias representativas de la diversidad viral a nivel mundial. Ambos linajes principales presentan amplia distribución mundial, aunque el linaje B reúne a la mayor parte de las secuencias de circulación actual.
A continuación se muestra una figura donde a la izquierda se encuentra un árbol filogenético construido a partir del alineamiento múltiple de 375 secuencias aisladas de pacientes de países de latinoamérica (GISAID), incluyendo secuencias argentinas, donde a su vez se indica el linaje a la cual corresponde cada secuencia. A la derecha se indica la procedencia geográfica donde fue obtenida cada una de las secuencias.
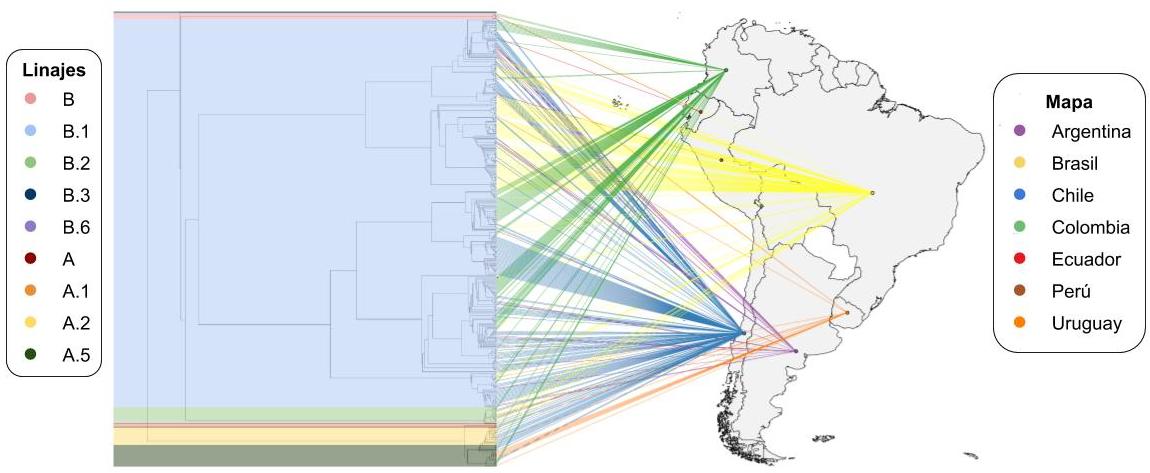
Figura 5: Árbol Filogenético de SARS-CoV-2 en Sudamérica. Las secuencias y coordenadas geográficas fueron obtenidas a partir de la metadata disponible en la base de datos GISAID a la fecha 12 de Junio, 2020. El árbol filogenético fue creado con IQtree (http://www.iqtree.org/) e integrado con el mapa a partir la biblioteca de R, phytools (http://www.phytools.org/). Cada secuencia está conectada por una línea coloreada con su país de procedencia (Referencia, cuadro izquierdo Mapa). Se analizó el linaje de las secuencias utilizando el programa Pangolin (https://pangolin.cog-uk.io/) y se las coloreó sobre las ramas del árbol filogenético (Referencia, cuadro derecho Linajes).
En la Figura 5 se puede observar qué linaje le fue asignado a cada una de las secuencias del árbol y de esta manera, teniendo en cuenta la temporalidad de la pandemia, nos permite hacer inferencias acerca del origen de los diferentes aislamientos. En el caso de las secuencias chilenas, podemos observar asignaciones a distintos linajes, incluidos algunos linajes “A.X” (circulantes en Reino Unido, EE.UU, España o Australia). Se pueden observar hasta 8 clusters de secuencias chilenas asignados a diferentes linajes, lo que podría indicar hasta 8 introducciones diferentes del virus generando clusters independientes de circulación local. No obstante, es de considerar que existen otras posibilidades para el caso chileno, como que hayan habido varias introducciones más del virus que no se puedan ver en la filogenia por falta de secuencias. También es importante tener en cuenta que la correlación entre países y linajes no se puede inferir de manera directa. Usualmente un mismo país presenta más de un linaje circulando, aunque suele haber un linaje preponderante.
En Argentina actualmente (junio de 2020) existen 29 secuencias de SARS-CoV-2 provenientes del AMBA, la ciudad de Bariloche y la ciudad de Bahía Blanca. En particular, las secuencias argentinas se asociaron, al menos, con cinco linajes internos: muchas corresponden al linaje B.1 (13), que es uno de los principales causantes de los brotes en Europa y América del Norte (https://github.com/hCoV-2019/lineages/), y en los otros casos se pudieron identificar linajes más específicos que no se detallan en la Figura 5, como B.1.1 (5), B.1.3 (3), B.1.5 (7) y B.1.27 (1).
¿Por qué es importante el estudio de las secuencias autóctonas de SARS-CoV-2?
Como se desprende de todo lo dicho anteriormente la secuenciación de genomas es una herramienta fundamental para analizar la entrada y circulación del virus. Este conocimiento, sin embargo, no solo nos permite establecer políticas de vigilancia sino adecuar la administración de futuras vacunas y antivirales para combatir cepas que circulan en nuestro país. Muchas investigaciones para el desarrollo de antivirales se basan en bloquear farmacológicamente la actividad de la proteína Mpro, esencial para la replicación del virus, y otras en la inhibición de la polimerasa viral (RdRp). Diferencias en el genoma viral pueden resultar en diferentes instrucciones de cómo fabricar estas proteínas y a su vez en proteínas resistentes a la terapia. De esta manera, conocer los diferentes genomas de coronavirus nos permite predecir variantes en estas proteínas, lo que es esencial para prever la eficacia de una posible terapia.
Estas mutaciones también nos permiten hacer distintos análisis acerca del diagnóstico y la virulencia del virus. Por ejemplo se puede verificar si algunas de las variantes circulantes en nuestro país se encuentran comprendidas en las regiones de apareamiento de los cebadores (“primers”) utilizados en los kits de diagnóstico por reacción en cadena de la polimerasa (PCR). En caso de que esto ocurra podría complicar el diagnóstico.
Otra variante que merece ser destacada por sus impactos en la infectividad de SARS-CoV-2 es la mutante D614G en la proteína Spike (S). El cambio de ácido aspártico (D) por una glicina (G) podría impactar en la posibilidad de transmisión viral pero no en la gravedad del cuadro clínico. Resultados experimentales in vitro han mostrado que la nueva cepa SG614 infecta con mayor eficiencia que la cepa SD614 y que se correlacionarían con los datos epidemiológicos [10][11]. Sin embargo, no se ha demostrado aún su relevancia in vivo. Estas mutaciones son características del linaje B y circulan con una muy alta prevalencia a nivel mundial, por lo que su presencia en Argentina confirma la persistencia de las mutaciones en el linaje en su introducción al país.
Perspectivas
Constituído por más de 100 investigadores de 30 instituciones de todo el país y del cual forman parte los autores de este trabajo, se conformó el Consorcio Argentino de Genómica de SARS-CoV-2, dirigido por la Dra. Mariana Viegas del Hospital Gutiérrez. Este Consorcio diseñó el Proyecto Argentino Interinstitucional de genómica de SARS-CoV-2 (PAIS, http://pais.qb.fcen.uba.ar/), financiado, a través del subsidio FONARSEC IP COVID-19 N° 247, por la Agencia Nacional de la Promoción de la Investigación, el Desarrollo Tecnológico y la Innovación del Ministerio de Ciencia, Tecnología e Innovación, Argentina. El objetivo de PAIS es el análisis de la trayectoria evolutiva de las cepas del SARS-CoV-2 que circulan en Argentina para estudiar su origen y dispersión en el país, en el contexto de las cepas mundiales, como así también analizar las mutaciones que pudieran afectar el diagnóstico, la transmisión y la virulencia del virus.
Para cumplir el objetivo se planea a corto plazo un muestreo en todo el país de aproximadamente 1000 genomas de SARS-CoV-2, para obtener sus secuencias genómicas, caracterizarlas y predecir el origen y esparcimiento de la pandemia sobre nuestro territorio. Así y a modo de síntesis, el contar con un número importante de secuencias genómicas de los virus que circulan en nuestro país resulta fundamental para pensar políticas de salud pública exitosas en el marco de la pandemia de SARS-CoV-2.
Referencias:
1. Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, et al. (2020) Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27: 325–328.
2. Dagur HS (2020) Genome organization of Covid-19 and emerging severe acute respiratory syndrome Covid-19 Outbreak: A Pandemic. Eurasian Journal of Medicine and Oncology doi:10.14744/ejmo.2020.96781
3. Chen Y, Liu Q, Guo D (2018) Emerging coronaviruses: Genome structure, replication, and pathogenesis. Journal of Medical Virology 92: 418–423.
4. Sosa EJ, Burguener G, Lanzarotti E, Defelipe L, Radusky L, Pardo AM, et al. (2018) Target-Pathogen: a structural bioinformatic approach to prioritize drug targets in pathogens. Nucleic Acids Research 46: D413–D418.
5. Shu Y, McCauley J (2017) GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveillance 22. doi:10.2807/1560-7917.ES.2017.22.13.30494
6. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. (2018) Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34: 4121–4123.
7. Rambaut A, Holmes EC, Hill V, O’Toole Á, McCrone JT, Ruis C, et al. (2020) A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. doi:10.1101/2020.04.17.046086
8. Holmes S (2003) Bootstrapping phylogenetic trees: Theory and Methods. Statistical Science 241–255. doi:10.1214/ss/1063994979
9. Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Systematic Biology 539–552. doi:10.1080/10635150600755453
10. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. (2020) Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. doi:10.1101/2020.04.29.069054
11. Zhang L, Jackson CB, Mou H, Ojha A, Rangarajan ES, Izard T, et al. (2020) The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. doi:10.1101/2020.06.12.148726
|
Revista QuímicaViva Número 2, año 19, Agosto 2020 quimicaviva@qb.fcen.uba.ar |