Critical assessment of structure prediction (CASP): 24 años evaluando predicciones de estructuras de proteínas
Luciano A. Abriata
Protein Purification and Structure Facility and Laboratory for Biomolecular Modeling, School of Life Sciences, École Polytechnique Fédérale de Lausanne and Swiss Institute of Bioinformatics. Laussane. Switzerland
Recibido: 04/12/2017 - Aceptado: 21/01/2018
Resumen
Sin dudas, la predicción de estructuras de proteínas es esencial en la investigación moderna en biología, ya que permite abordar cuestiones estructurales de proteínas difíciles de trabajar en el laboratorio o inclusive de proteínas que son accesibles experimentalmente pero para las cuales el detalle requerido no justifica el tiempo y costo de experimentos. Es crítico, sin embargo, contar con evaluaciones y un marco de referencia sobre la calidad de las predicciones, las posibilidades, y las limitaciones. La Critical Assessment of Structure Prediction (CASP) se enfoca precisamente en ese objetivo de evaluar métodos, servidores y grupos de investigadores dedicados a predecir estructuras de proteínas. CASP se organiza en un formato de competición bianual donde los organizadores colectan estructuras que no han sido publicadas y dan a los predictores participantes sus secuencias aminoacídicas. Los predictores luego generan y envían sus modelos, y un grupo independiente de asesores los evalúa. Luego de prácticamente un cuarto de siglo desde la primera edición de CASP, este artículo introduce en forma sencilla las principales observaciones de la edición número 12 (2016-2017), resumiendo el progreso registrado, los métodos y grupos de predictores más destacados, el estado del arte de modelado, y las limitaciones actuales.
Palabras clave: modelado, proteínas, coevolución, predicción, bioinformática
24 years of evaluation of protein modeling strategies by the Critical Assessment of Structure Prediction contest
Summary
The ability to predict protein structures is essential in modern biology, as it allows researchers to approach structural details of proteins that are hard to work with in the wet lab, or even of proteins amenable to wet lab experimentation but where the questions at stake do not justify the costs and time required to solve the structure experimentally. It is critical, however, to have objective evaluations and reference frames about the accuracy of structure predictions, their potentials and limitations. The Critical Assessment of Structure Prediction (CASP) focuses on this aim of providing objective evaluations of methods and research groups for protein structure prediction. Every two years, CASP organizers secure new protein structures that have not been released to the public, provide predictors with their amino acid sequences, collect the models submitted by them, and then independent groups of assessors evaluate the submitted models along different tracks (here only the tertiary structure prediction track is reviewed). After roughly a quarter century of CASP, this article recaps in a lay manner the most important observations during CASP round 12 (2016-2017), summarizing progress over time and highlighting the most successful methods and groups, the state-of-the-art of modeling strategies, and current potential and limitations.
Keywords: modeling, proteins, coevolution, prediction, bioinformatics
Introducción
La biología estructural intenta explicar los sistemas biológicos a nivel atómico. Para esto, la disciplina depende críticamente de la disponibilidad de estructuras de las moléculas involucradas, entre las cuales las más importantes suelen ser proteínas. Mientras que muchas estructuras pueden determinarse mediante técnicas experimentales como difracción de rayos X o neutrones, resonancia magnética nuclear, y ahora también crio-microscopía electrónica, existe también la alternativa de predecir, o “modelar”, estas estructuras usando métodos computacionales y a menudo algo de “intuición química” y datos estructurales de baja resolución.
Naturalmente, tales predicciones son esenciales para un gran número de moléculas biológicas que no se pueden producir en las cantidades y condiciones necesarias para los distintos experimentos. Pero predecir estructuras puede ser también útil para casos de moléculas que quizás no sean difíciles de producir y de manipular durante los experimentos requeridos para resolver estructuras, pero para las cuales la cantidad de información provista por la estructura no justifique los costos y tiempos. De hecho, si pudiéramos predecir estructuras de biomoléculas con suficiente confianza, podríamos enfocar los experimentos sólo en sistemas particularmente difíciles (esto es lo que de hecho persiguen los esfuerzos de genómica estructural) o en estudiar efectos de perturbaciones en la estructura, como por ejemplo el efecto de la unión de un ligando a la proteína en estudio. En el extremo, si pudiéramos predecir toda la fisicoquímica de un dado sistema a nivel atómico, podríamos prescindir totalmente de experimentos para la determinación de estructuras, y podríamos concentrar los esfuerzos directamente en entender mecanismos y todo lo que su conocimiento permite hacer: desarrollo de drogas, diseño de nuevas funciones, entender la evolución, etc.
Dado el impacto que pueden entonces tener las predicciones de estructuras sobre la biología estructural, generaciones de investigadores han trabajado en el problema desde mediados del siglo pasado, especialmente para proteínas ya que poseen un valor y una variedad estructural mucho mayores que otras macromoléculas biológicas. Muchísimos métodos han sido desarrollados, los cuales se pueden clasificar en dos grandes grupos. Por un lado, aquellos que utilizan estructuras ya conocidas para intentar predecir estructuras de proteínas de secuencia o plegamiento similar, lo cual se conoce como “modelado por homología”. Por otro lado,aquellos métodos que intentan “plegar” secuencias independientemente de cualquier homología con otras proteínas de estructura conocida, por ejemplo utilizando simulaciones basadas en principios fisicoquímicos básicos o utilizando información sobre la estructura de pequeños fragmentos de péptidos y/o de contactos entre residuos.
CASP: Evaluando métodos y grupos para predicción de estructuras de proteínas
El gran problema que surgió junto con los métodos, programas y expertos en modelar estructuras, es como evaluar la calidad de estas predicciones. A principios de los años 1990 nació la Critical Assessment of Structure Prediction, o CASP, una organización cuyo objetivo es proveer un seguimiento y evaluación constante de los métodos disponibles para predecir estructuras de proteínas [1]. La competición tiene lugar cada 2 años, durante los cuales los organizadores colectan estructuras experimentales (“targets”) que no han sido publicadas en el Protein Data Bank. Los organizadores proveen las secuencias de aminoácidos de estas proteínas a los grupos predictores, quienes luego de un período de tiempo determinado envían sus predicciones a los organizadores. Luego un grupo de asesores (independiente de la organización y que no participan como predictores) evalúa los modelos provistos por los predictores en comparación con las estructuras experimentales (a la que sólo los asesores tienen acceso). Cada competencia finaliza con una serie de artículos que describen la dificultad presentada por los targets, describen la calidad de los modelos proporcionados por los predictores, generan un ranking “oficial” de los predictores, y discuten el “estado del arte” del modelado, especialmente qué métodos funcionaron, qué cuestiones estructurales fueron especialmente difíciles de predecir, etc. En todas las ediciones de CASP esta fue la competición principal, pero en cada edición hay otras vías que evalúan distintos aspectos, como predicción de complejos, refinamiento de detalles, predicción de función, etc.
En 2016 y 2017 tuvo lugar la CASP12, para la cual este autor y el grupo al que pertenece fueron asesores en la vía principal de la competición, enfocada en la predicción de estructuras terciarias para targets difíciles. En lo que sigue se describen las cuestiones más interesantes derivadas de esta experiencia, mientras que el lector puede acudir a las publicaciones principales [2,3] (open access) para obtener todos los detalles.
Estado del arte del modelado de proteínas con dominios clasificados como difíciles en CASP12
Normalmente, los targets disponibles en cada CASP se fraccionan en unidades de evaluación según varios criterios basados en métricas automáticas y en el análisis visual de las estructuras del target, de los posibles moldes (“templates”) disponibles en el Protein Data Bank, e inclusive de los modelos suministrados por los predictores. Las unidades de evaluación luego se clasifican de acuerdo a su dificultad; en CASP12 las clases fueron TBM, por “template-based modeling” las cuales deberían ser fáciles de predecir ya que existen buenos moldes en el Protein Data Bank), FM por “free modeling” significando que no hay moldes obvios, y FM/TBM que agrupa las unidades de evaluación para las cuales hay moldes similares ya sea a nivel de estructura o de secuencia, pero no ambas, o para las cuales a pesar de existir buenos moldes, las predicciones son de mediana calidad. En total, en CASP12 hubo 39 unidades de evaluación clasificadas como TBM, 19 como FM/TBM, y 38 como FM [2]. Las unidades FM y FM/TBM fueron evaluadas por un grupo de asesores que incluyó a este autor, donde el objetivo principal es determinar primero la calidad del plegamiento general y, en el caso de modelos que capturan el plegamiento muy bien, evaluar detalles más finos [3]. Las unidades de evaluación TBM son sujetas a análisis distintos que se enfocan específicamente en detalles, a cargo de otros asesores[4].
Las unidades de evaluación de CASP12 estuvieron entre los más difíciles en la historia de la CASP [2]. Para muchas no existen estructuras de proteínas homólogas en el Protein Data Bank (que los predictores pudieran utilizar para modelarlas); mientras que en algunos casos existen proteínas de plegamiento similar pero logrado mediante una secuencia totalmente distinta, lo cual dificultó a los predictores encontrarlas mediante búsquedas a nivel de secuencias. Fue reconfortante observar que para muchas de estas unidades de evaluación difíciles, hubo al menos un modelo de muy buena calidad. De hecho, anticipando un análisis del progreso en CASP, esta fue una ronda muy satisfactoria con un incremento importante en la calidad global de las predicciones respecto a ediciones previas.
Ejemplos de predicciones en CASP12
En CASP12 se utilizaron varios scores para evaluar objetivamente los modelos y guiar la evaluación visual por parte de los asesores, que es siempre esencial. El más importante de estos scores es el llamado GDTTS, por Global Distance Test-Total Score, que mide la fracción de residuos del modelo que pueden ser alineados (a nivel de su carbono alfa) con los correspondientes residuos en la estructura target dentro de los 1, 2, 4 y 8 Å. De esta forma, GDTTS captura tanto características globales del plegamiento como detalles finos, adoptando valores de ~10-20 para modelos totalmente incompatibles con el target hasta 100 para modelos cuyos residuos encajan todos perfectamente dentro de 1 Å de distancia del target, es decir esencialmente idénticos al target.
A continuación se describen brevemente los resultados para 6 unidades de evaluación interesantes, representativas de algunos de los casos encontrados. El lector interesado puede consultar el artículo oficial de CASP (open access) para conocer más detalles sobre la evaluación de modelos para todas las unidades, teniendo en cuenta varios scores además de GDTTS [3]. Además, todos las predicciones pueden ser consultadas en 3D interactivo en la siguiente web app [5,6] introducida en CASP12 para facilitar la evaluación y abrirla a la comunidad en forma transparente: http://predictioncenter.org/casp12/FM_assessors_app.html
T0866 (Figura 1A) es un caso de un target difícil para el cual muchos grupos de predictores produjeron buenos modelos. T0866 es un homohexámero del cual esta vía de la CASP sólo evaluó el monómero (otra vía se encargó de evaluar oligómeros), que constituye una sola unidad de evaluación. Comparado con los mejores posibles templates disponibles en el Protein Data Bank, el target tiene un plegamiento que guarda ciertas semejanzas, pero con numerosas inserciones y deleciones de tamaño importante. A pesar de ser un target difícil, la distribución de GDTTS muestra alrededor de 90 modelos con GDTTS entre ~70 y 81, es decir muy buenos. Los varios modelos de GDTTS > 80 son todos bastante parecidos entre ellos, capturando muy bien el corazón globular y mostrando diferencias entre sí y con el target principalmente en un bucle de unos 10 residuos.Debido al gran número de buenos modelos, no parece surgir ningún “ganador” claro para T0866.
Posibles razones del éxito en el modelado de T0866 incluyen la presencia de múltiples moldes, aunque todos con numerosas inserciones y baja similitud de secuencia. Pero además, se advierte para este target un gran número de secuencias homólogas que permitieron a algunos predictores aplicar una nueva serie de métodos útiles para la predicción de estructuras, basados en predecir contactos entre pares de residuos a partir de sus velocidades de coevolución. De hecho, una ejecución “naïve” (es decir sin ninguna intervención ni optimización) de la secuencia de este target en el servidor EVFold [7,8] para modelado de proteínas mediante contactos predichos por coevolución, resulta en un modelo con GDTTS de 39.7, es decir en el medio de la distribución de GDTTS de los modelos provistos por los predictores. Tal modelo es de calidad suficiente como para capturar la forma global del target (Figura 4).
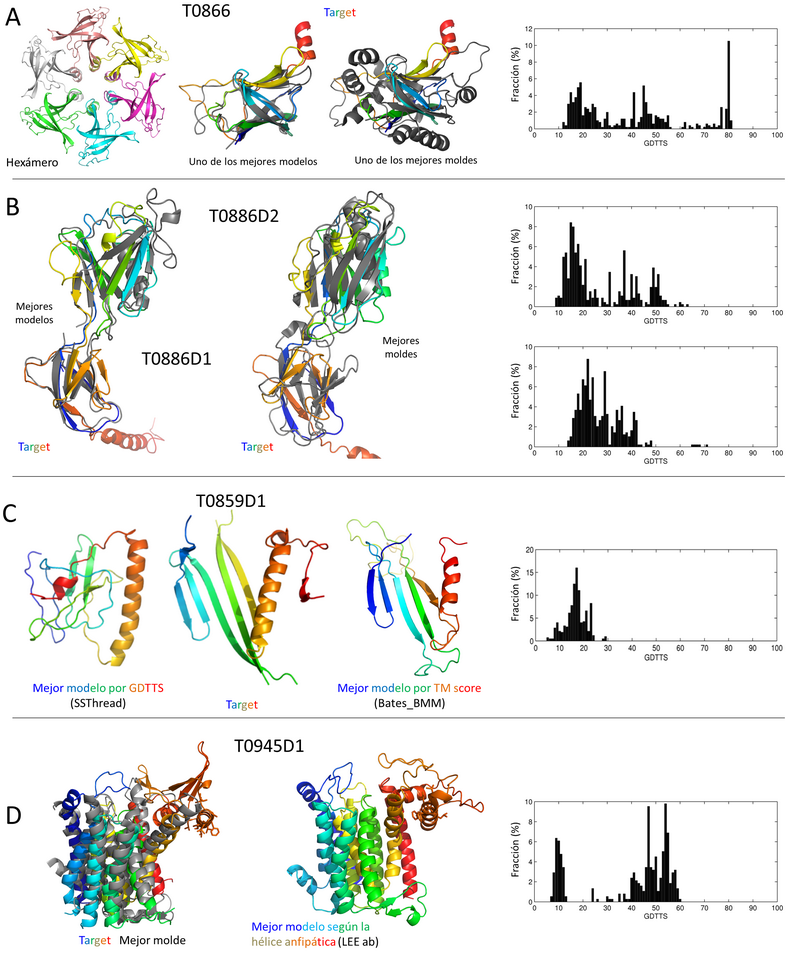
Figura 1: Resultados para las unidades de evaluación T0866 (A), T0886D1 y D2 (B), T0859(C) y T0945 (D). Para cada una se muestra la estructura target, los mejores modelos, los mejores moldes en los casos relevantes, y un histograma de los valores de GDTTS para todos los modelos enviados por los predictores.
T0886 (Figura 1B) es otro caso difícil, ejemplo de aquellos donde un grupo se destaca en especial. T0886 se evaluó como dos unidades, T0886D1 y T0886D2. La primera es discontinua en secuencia, es decir que está formada por dos segmentos de la secuencia separados entre sí por un segmento que corresponde a la segunda unidad.
Para T0886D1, las 5 predicciones del grupo de Baker tienen GDTTS entre 65 y 71, mostrando buena similitud global del plegamiento al compararlos con el target. Todos los otros modelos tienen GDTTS menor a 50, indicando que los modelos del grupo de Baker son sustancialmente mejores. Notablemente, los modelos de Baker capturan bien la discontinuidad de secuencia, lo cual ellos lograron mediante predicción de contactos a partir de medidas de coevolución en un gran alineamiento. Tal predicción de contactos muestra claramente una organización con un dominio discontinuo, que permitió al grupo de Baker encontrar buenos moldes de proteínas que tenían un plegamiento similar pero logrado mediante segmentos continuos de secuencia.
También para T0886D2, los 5 modelos del grupo de Baker fueron los mejores, aunque en este caso los modelos propuestos por otros grupos no estaban tan lejos en calidad.
T0859 (Figura 1C) es un ejemplo de un target con predicciones de mediana calidad. La dificultad en este target surge de que a pesar de existir en el Protein Data Bank una estructura similar, esta no guarda ninguna semejanza con el target al nivel de secuencia, con lo cual resultó prácticamente imposible para los predictores encontrarla, salvo para el grupo de Bates. Es importante que en este caso, búsquedas en base de datos de secuencias no encuentren otras secuencias con las cuales los predictores pudieran armar alineamientos para poder avanzar el modelado, como si fue el caso para T0866 y T0886 descriptos arriba.
Los mejores modelos para T0859 resultan los de Bates, basados en el molde de estructura similar, y los del grupo SSThread. Ambos capturan la segregación de una alfa hélice respecto a una hoja beta, necesaria para un cambio de dominio que se verifica en el ensamblado biológico tanto del target como del modelo. Sin embargo, varios detalles inclusive el registro de las hojas beta son incorrectos, con lo cual todos los scores usados son relativamente bajos.
T0945 (Figura 1D) es una proteína de membrana que consiste en una sola unidad de evaluación, del tipo FM/TBM. La distribución de GDTTS muestra un grupo de predicciones razonables a buenas con GDTTS entre 40 y 60. Los modelos con GDTTS cercano a 60 muestran una muy buena región transmembrana, excepto en un pequeño bucle y en una inserción de unos 80 residuos. Esta última forma en el target una pequeña hoja beta con una alfa hélice anfipática que, dada su disposición, muy probablemente se recuesta sobre la superficie de la membrana en ese entorno. La mayoría de los grupos empacaron esta hélice contra el resto de la proteína, escondiendo su región hidrofóbica. Sólo dos grupos (Leey Zhang) propusieron modelos donde la hélice adopta una posición similar a la observada en el target, pero rotada de forma tal de esconder sus aminoácidos hidrofóbicos, con lo cual no capturaron su principal característica.
T0896 (Figura 2) es un ejemplo de aquellos targets para los cuales no hay buenas predicciones, especialmente si consideramos su unidad de evaluación T0896D3. Esta unidad es de la clase FM, ya que no hay proteínas con secuencia ni estructura similar en el Protein Data Bank. Encima, posee 80% de sus residuos en una conformación cristalográficamente definida pero sin estructura secundaria regular, y está dispuesto alrededor de la unidad de evaluación T0896D2. Las predicciones para T0896D3 son todas muy pobres, y de hecho la inspección visual no revela ningún modelo que capture al menos alguna característica gruesa. A su vez, la dificultad en modelar T0896D3 complica el modelado de las unidades T0896D1 y T0896D2, que deberían ser relativamente fáciles ya que existen moldes de secuencia y estructura similares en el Protein Data Bank.
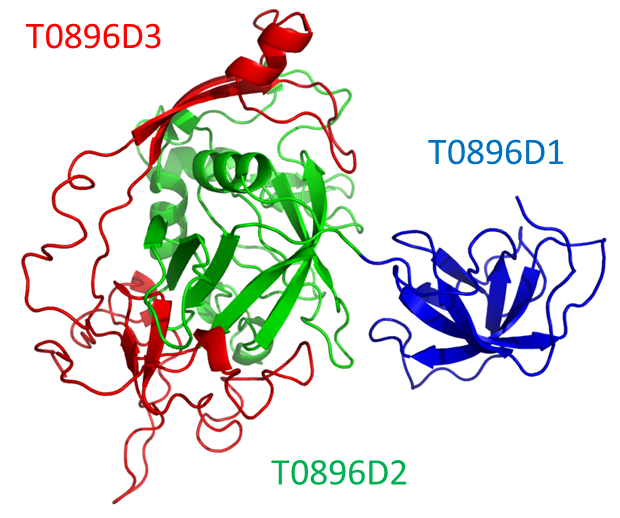
Figura 2: Target T0896, con sus 3 unidades de evaluación en distintos colores. Nótese como T0896D3 contiene una gran porción sin estructura definida (80%) que se envuelve alrededor de T0896D2.
Progreso, razones del progreso, y limitaciones actuales
A pesar de la alta dificultad de muchos targets de CASP12, la evalución de los modelos indicó muchas buenas predicciones, a veces por varios grupos, a veces por grupos específicos. En particular, los grupos de Baker, Lee, Zhang, y el conjunto WeFold, propusieron muchos modelos buenos para varias unidades de evaluación.
Una pregunta importantísima en la comunidad de la CASP es cuanto progreso se observa realmente en función del tiempo. Esta es una pregunta compleja porque depende de la dificultad de los targets presentes en cada edición, de la cantidad de información y moldes disponibles para ellos, e inclusive de algunas cuestiones subjetivas sobre la evaluación de las unidades de evaluación e inclusive sobre su definición. Una primera aproximación utilizando los scores GDTTS de los mejores modelos contribuidos en cada CASP, promediados como la mediana con su desviación en la Figura 3A, muestra que entre CASP1 y CASP5 hubo una rápida mejora en los métodos de modelado de proteínas, seguido de una situación estable hasta CASP11 y luego un pequeño salto positivo de CASP11 a CASP12. Esto es interesante y sugiere que los métodos han evolucionado y/o incorporado nuevas fuentes de datos.
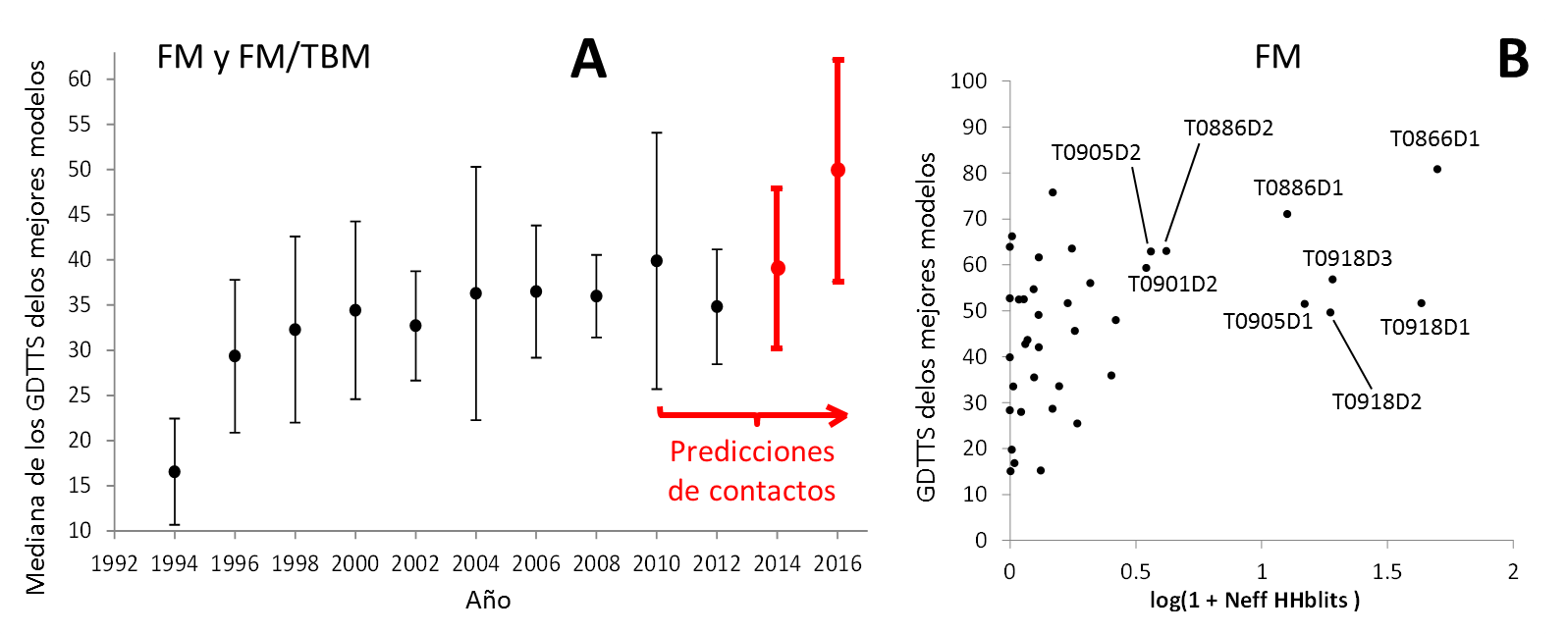
Figura 3: (A) Progreso en CASP, medido como la mediana de los valores de GDTTS de los mejores modelos para todas las unidades de evaluación difíciles (FM y FM/TBM). Las barras rojas indican las 2 últimas ediciones de CASP (11 y 12). (B) GDTTS del mejor modelo para cada unidad de evaluación, graficado contra el número de secuencias obtenidas con HHblits para cada unidad normalizada por su largo en número de residuos (Neff, graficado como el logaritmo desplazado en 1).
Como se mostró en algunos casos y se puede explorar en más detalle en el artículo original de la evaluación de CASP12 así como en literatura reciente, los métodos y grupos predictores más modernos están incorporando predicciones de contactos entre residuos para asistir sus predicciones de estructuras (Figura 4 ejemplificando con el target T0866). Estos métodos se basan principalmente en cálculos de coevolución de todos los pares de residuos posibles en una dada secuencia. Básicamente, la idea subyacente es que pares de residuos que están en contacto en la estructura tridimensional deben sufrir sustituciones en forma correlacionada durante la evolución, de forma tal de preservar su interacción. Entonces, dado un alineamiento, estos métodos descomponen las sustituciones de aminoácidos en cada posición de la secuencia y calculan cuánto co-evolucionan todos los pares de residuos. En CASP12 observamos que entre las unidades de evaluación más difíciles (FM), aquellas para las cuales se pueden armar alineamientos más profundos (es decir con más secuencias por residuo) logran obtener mejores modelos (Figura 3B). No es así para las unidades de evaluación clasificadas como FM/TBM y TBM, donde probablemente los grupos y programas predictores prefirieron utilizar métodos más convencionales basados en modelado por homología. En resumen, esto indica que en el estado del arte, para proteínas para las cuales no hay otras homólogas de estructura conocida, es todavía posible derivar modelos razonables mediante predicciones de contacto, al menos si es posible encontrar un número suficiente de secuencias homólogas. Esto abre una nueva puerta en el mundo del modelado de proteínas, que se ampliará a medida que los proyectos de secuenciación permitan armar alineamientos más grandes y a medida que los métodos de predicción de contacto mejoren (al respecto, el lector puede también referirse al artículo que evaluó predicciones de contactos en CASP12 [9]).
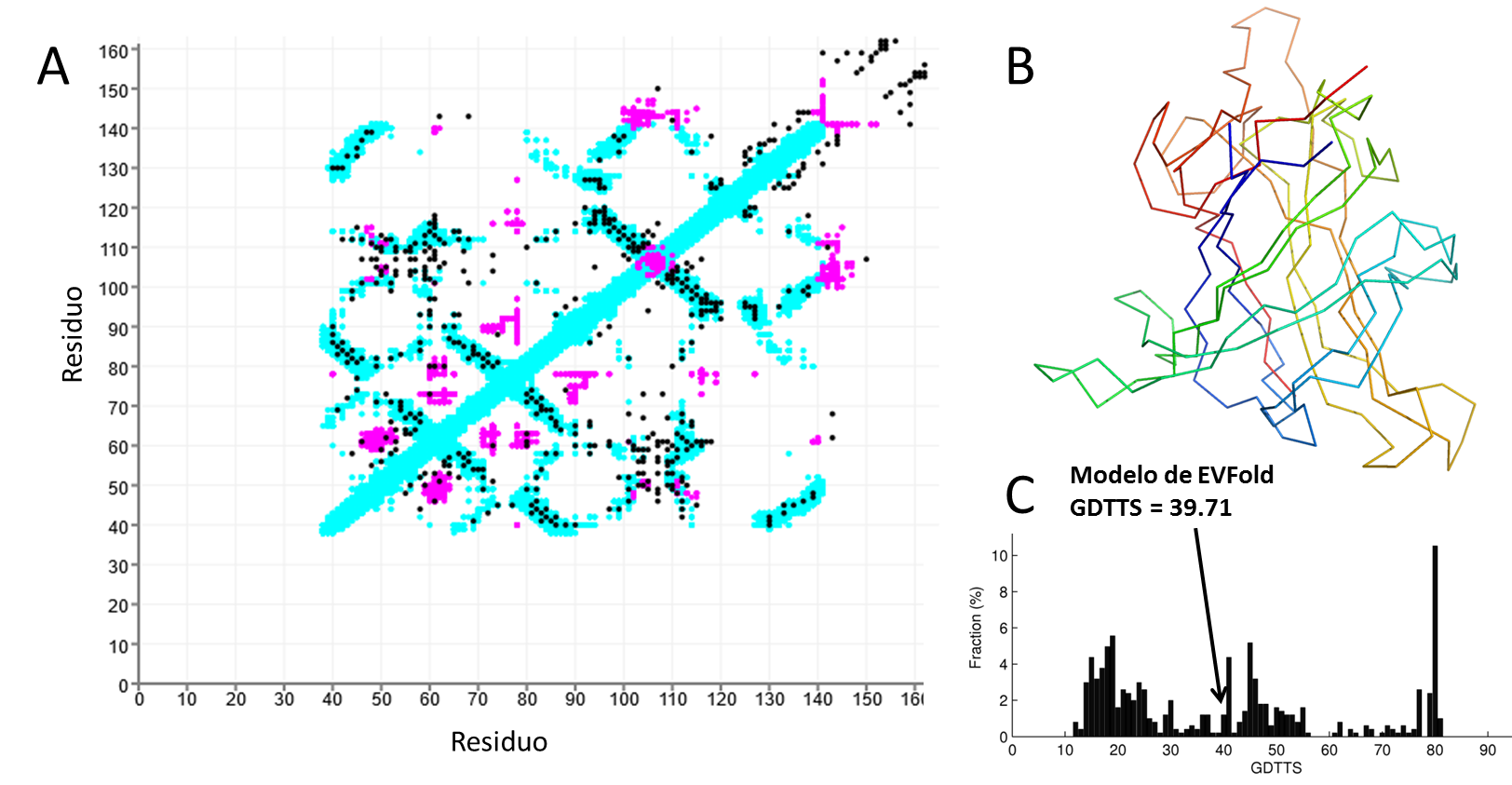
Figura 4: (A) Mapa de contactos entre residuos calculado a partir de la estructura cristalográfica del target T0866D1 dentro del monómero (celeste) y entre monómeros adyacentes (magenta), sobre el cual se marcan los residuos predichos en contacto por EVFold desde medidas de coevolución. (B) El modelo calculado por EVFold para T0866 a partir de los contactos predichos, alineado en 3D con la estructura conocida. (C) Distribución de valores de GDTTS para todos los modelos provistos por predictores para T0866D1, con una flecha que indica donde cae la predicción “naïve” de EVFold.
Limitaciones actuales
El análisis de las predicciones permitió también determinar cuáles son algunos de los problemas importantes, que ni métodos automatizados ni humanos expertos pudieron resolver.
Proteínas de más de 150 residuos son muy difíciles de modelar cuando no existen homólogos de estructura conocida para usarlos como molde ni suficientes secuencias como para predecir contactos en forma confiable. Por el contrario, varias proteínas de menos de 125 residuos pudieron ser predichas al menos globalmente, aun en casos donde no había homólogos de estructura conocida y unas pocas secuencias para predecir contactos.
Un problema recurrente es la existencia de homólogos estructurales sin similitud de secuencia, de forma tal que el molde pasa inadvertido y dificultando el modelado del target, como en T0859. La futura introducción de nuevos métodos para detectar similitud de secuencias, por ejemplo comparando propiedades fisicoquímicas [10], seguramente ayudará en este aspecto. También, algunos grupos utilizaron en CASP12 predicciones de contactos desde secuencias para derivar modelos de topología que permitieran encontrar moldes adecuados, y luego modelar el target en base al molde encontrado [11]. Por otro lado, aún en casos donde es posible encontrar moldes, resulta claro que eliminar inserciones del modelo respecto al target es mayormente satisfactoria, pero lo contrario, es decir la inserción de fragmentos del target inexistentes en el molde, suele resultar dificultosa. Otro problema esla falta de secuencias suficientes no sólo para calcular contactos entre residuos sino también para poder encontrar homólogos de estructura conocida pero poca similitud de secuencia.
Desde el punto de vista de las estructuras, problemas importantes son la existencia de “domain swaps”, es decir segmentos de secuencia que se intercambian con dominios adyacentes; bucles largos e dominios con un gran número de residuos sin adoptar una estructura secundaria, los cuales complican también el modelado de dominios adyacentes como en T0896; también dominios discontinuoscomo en T0886 descripto arriba. La oligomerización en algunos casos complica el modelado; y en proteínas de membrana, si bien los métodos son medianamente confiables para el corazón transmembrana, cuestiones detalladas como la hélice anfipática de T0945 son complicadas.
Conclusiones
La Critical Assessment of Structure Prediction (CASP), con su edición bianual número 12 recién terminada, provee a la comunidad de desarrolladores y usuarios de métodos de modelado de proteínas una evaluación constante del estado del arte. Esta última edición reveló una mejora importante en el modelado de sistemas difíciles, en gran parte debida a la introducción de métodos para la predicción de contactos entre residuos,que sirven para definir dominios, asistir la búsqueda de moldes, y en algunos casos directamente modelar la estructura. Las próximas ediciones de CASP mostrarán si estos métodos pueden mejorar aún más; probablemente lo hagan, pero además, la ampliación de las bases de datos de secuencias seguramente permitirá ampliar la aplicación de estos métodos a más proteínas.
Referencias:
1. Bourne PE (2003) CASP and CAFASP experiments and their findings. Methods of Biochemical Analysis 44: 501–507
2. Abriata LA et al (2017) Definition and classification of evaluation units for tertiary structure prediction in CASP12 facilitated through semi-automated metrics. Proteins DOI: 10.1002/prot.25403
3. Abriata LA et al Assessment of hard target modeling in CASP12 reveals an emerging role of alignment-based contact prediction methods. Proteins: Structure Function and Bioinformatics. DOI: 10.1002/prot.25423
4. Kryshtafovych A et al (2017) Evaluation of the template-based modeling in CASP12. Proteins DOI: 10.1002/prot.25425
5. Abriata LA (2017) Web Apps Come of Age for Molecular Sciences. Informatics 4,
6. Abriata LA et al (2017) Augmenting research, education and outreach with client-side web programming. Trends in Biotechnology DOI: https://doi.org/10.1016/j.tibtech.2017.11.009
7. Marks DS et al (2012) Protein structure prediction from sequence variation. Nature Biotechnoogy 30: 1072–1080
8. Marks DS et al (2011) Protein 3D structure computed from evolutionary sequence variation. PloS One 6, e28766
9. Schaarschmidt J et al (2017) Assessment of contact predictions in CASP12: co-evolution and deep learning coming of age. Proteins DOI: 10.1002/prot.25407
10. He Y et al (2015) Alternative approach to protein structure prediction based on sequential similarity of physical properties. Proceedings of the National Acadademy of Sciences of the United States of America 112: 5029–5032
11. Ovchinnikov S et al (2017) Protein structure prediction using Rosetta in CASP12. Proteins DOI: 10.1002/prot.25390
|
Revista QuímicaViva Número 1, año 17, Abril 2018 quimicaviva@qb.fcen.uba.ar |