Circuitos inmunológicos y vasculares mediados por lectinas y glicanos: implicancias en el desarrollo de nuevas terapias en cáncer y enfermedades autoinmunes
Nicolás A. Pinto1#, Luciano G. Morosi1#, Sebastián M. Maller1#, Gabriel A. Rabinovich1,2
1Laboratorio de Inmunopatología, Instituto de Biología y Medicina Experimental (IBYME), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET). 2 Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Buenos Aires, Argentina.
#N.A.P., L.G.M. y S.M.M. contribuyeron en forma equivalente a este trabajo
Resumen
Las glicoproteínas presentes en la superficie celular, como así también las que componen la matriz extracelular, contienen información vital codificada en glicanos. Estos glicanos son ensamblados y modificados post-traduccionalmente por distintas enzimas llamadas glicosiltransferasas y glicosidasas presentes en el lumen del Retículo Endoplásmico (RE) y en el Aparato de Golgi. Dado que la biosíntesis de los mismos se efectúa sin ningún tipo de molde, existe una enorme diversidad de estructuras sacarídicas presentes en proteínas y lípidos. Esto determina que el estudio de la información contenida en los distintos glicanos sea un verdadero desafío para los glicobiólogos en general, y para los glicoinmunólogos en particular. Sin embargo, en las últimas décadas se produjo un avance significativo en la comprensión de los “glico-códigos” presentes en la superficie celular y en proteínas que se encargan de interpretar y decodificar esta información, denominadas lectinas. En esta revisión nos concentraremos en las galectinas, una familia de lectinas solubles secretadas al medio extracelular por una vía no clásica, definidas por su capacidad de reconocer residuos β-galactósidos, presentes en glicoconjugados expresados en una gran variedad de tipos celulares. Dada la capacidad de algunas galectinas de comportarse como ligandos multivalentes (en condiciones particulares de concentración, pH y estado de oxidación), estas proteínas pueden entrecruzar o segregar receptores de membrana glicosilados modulando así vías de transducción de señales gatilladas por factores presentes en el microambiente celular. A su vez, estas lectinas pueden comportarse como ligandos no canónicos de una variedad de receptores de importancia en programas inmunológicos y vasculares. Esta regulación de vías de señalización conlleva a varios tipos de respuestas biológicas asociadas a la activación, la adhesión, la migración, la proliferación y la muerte celular. En particular, focalizaremos nuestra atención en galectina-1 (Gal-1), un miembro prototípico de esta familia, como regulador homeostático de la respuesta inmunológica y como factor pro-angiogénico, y su implicancia en el desarrollo y resolución de enfermedades autoinmunes y neoplásicas, enfatizando en aportes realizados por nuestro grupo de investigación.
Palabras clave: Galectinas, glicanos, glicosilación, inmunidad, cáncer, autoinmunidad.
Regulatory circuits mediated by lectin-glycan interactions in immune and vascular signaling programs: Implications in the design of novel therapeutic strategies in autoimmune diseases and cancer
Summary
Glycoproteins present at the cell surface or within the extracellular matrix are composed of a myriad of glycans assembled through a post-translational modification process called glycosylation. These molecules store critical biological information that controls cellular activation, differentiation and survival, and their synthesis is controlled by the sequential action of a limited number of glycosyltranferases and glycosidases located in the endoplasmic reticulum (ER) and Golgi apparatus. Since the glycosylation process is not template-driven, there are almost infinite possibilities of saccharide linkages and structures which hindered the progress of functional glycobiology for many decades. However, identification of glycan-binding proteins or lectins responsible of deciphering the cellular ‘glyco-codes’, has illuminated new insights into our understanding of the functions of glycans in a broad range of cellular processes in health and disease. In this review we will focus on galectins, a particular family of soluble lectins which are secreted to the extracellular medium through a non-conventional ER-Golgi-independent route. Galectins are defined based on their ability to bind β-galactosidase residues in glycoproteins and glycolipids. Given the ability of some members of the galectin family to form multivalent structures (depending on their concentration, redox and pH status), they are able to control clustering, reorganization and trafficking of relevant glycosylated receptors. These multivalent complexes can in turn regulate receptor signaling, endocytosis and activation within immune and vascular compartments. Galectin-mediated regulation of receptor signaling results in modulation of a myriad of cellular processes including cell adhesion, migration, proliferation and apoptosis. Our results during the past decade demonstrated that galectin-1 (Gal-1) controls immune and/or vascular homeostasis through multiple regulatory mechanisms. Here we will discuss pioneer and recent studies highlighting the role of Gal-1 as a novel target in cancer, inflammation and autoimmunity.
Keywords: Galectins, glycans, glycosylation, immunity, cancer, autoimmunity.
Glicanos y Lectinas
Todas las células, desde las bacterias hasta las neuronas o linfocitos, poseen estructuras sacarídicas (glicanos) unidas a proteínas y/o a lípidos presentes en la superficie celular. Estos glico-conjugados se ensamblan de manera secuencial, sin un molde pre-establecido, mediante la acción concertada de glicosiltransferasas y glicosidasas. Los glicanos poseen roles cruciales en varios procesos biológicos en organismos superiores: migración celular, diferenciación, comunicación celular, inmunidad y vascularización [1–4]. En el sistema inmunológico, particularmente, regulan la activación, diferenciación y homeostasis de células encargadas de defendernos de posibles agentes agresores [5].
Las dos principales formas de glicosilación en proteínas (N- y O-glicosilación, Figura 1) lejos de ser fenómenos estáticos, consisten en procesos dinámicos que dependen del tipo de célula y su estado fisiológico de activación/diferenciación, como así también de señales del microambiente en el cual se encuentran, de la expresión y/o control epigenético de diversas glicosiltransferasas y glicosidasas, de la distribución subcelular de componentes de la maquinaria de glicosilación, del estado nutricional de las células, de la disponibilidad de azúcares nucleótidos, de los niveles de oxígeno del medio y de factores de crecimiento, y citoquinas. Procesos patológicos como inflamación crónica y cáncer también generan cambios en los perfiles de glicosilación celular en los tejidos involucrados [6–10]. Dada la variedad de posibles estructuras de glicanos que pueden generarse, los mismos no representan simples “ornamentos” de la superficie celular (como durante mucho tiempo se especuló) sino que proveen información valiosa que determina el desarrollo de procesos fisiológicos y patológicos.
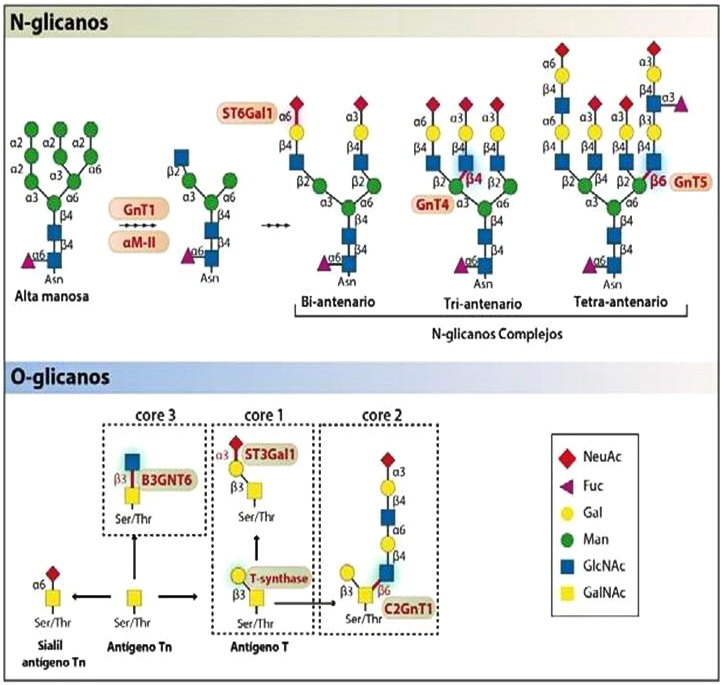
Figura 1: Vías de glicosilación involucradas en la síntesis de ligandos de galectinas. El esquema muestra simplificados los dos tipos de glicosilación principales sobre proteínas, la N- y la O-glicosilación. La N- glicosilación consiste en la adición de glicanos en un residuo de asparagina de una proteína alrededor de una región consenso –N-X-S/T (X no puede ser prolina), mientras que la O-glicosilación ocurre más tardíamente en el Golgi, sobre los aminoácidos serina y treonina. El proceso de N- glicosilación comprende la modificación secuencial de un glicano inicial básico rico en manosas por parte de distintas glicosidasas y glicosiltransferasas. Durante el proceso de formación de N glicanos complejos, es crítica la actividad secuencial de las enzimas N-acetilglucosaminiltransferasas (Mgat1; 2, 4 y 5), las cuales generan las ramificaciones de GlcNac que constituyen los ligando preferidos por las galectinas. Por otro lado, otra de las enzimas importantes es la α2,6 sialiltransferasa 1 (ST6Gal1), la cual incorpora ácido siálico en posición α2,6 inhibiendo la unión de ciertas galectinas sobre N-glicanos complejos. Con respecto a la O-glicosilación las enzimas más relevantes son la T-sintasa esencial para la biosíntesis de core-1-O-glicanos y la α2,3 sialiltransferasa (ST3Gal1), encargada de agregar ácido siálico en core-1O-glicano.
Las moléculas encargadas de decodificar la información biológica encriptada en estos glicanos, son las lectinas, proteínas endógenas capaces de reconocer estructuras glicosídicas complejas presentes en glicoproteínas y glicolípidos, las cuales al formar complejos específicos gatillan respuesta celulares que depende de la naturaleza de estas lectinas y de los glicanos con los cuales interactúan [11, 12]. Diversos estudios, utilizando modelado molecular han demostrado que las interacciones lectinas-glicanos conducirían a la formación de estructuras tridimensionales complejas denominadas “entramados” (lattices, en inglés) [12], capaces de organizar dominios supramoleculares en la membrana plasmática, que pueden segregar glicoproteínas y modificar el umbral de señalización, activación y endocitosis de receptores relevantes [13, 14]. En muchos casos estos complejos pueden sustituir a ligandos canónicos y activar receptores desencadenando respuestas biológicas críticas como activación, diferenciación y muerte celular [13, 14].
Se han caracterizado tres familias importantes de lectinas involucradas en procesos de reconocimiento y señalización en el sistema inmune, como así también en programas vasculares: a) Receptores lectina de tipo C (C-type lectin receptors, CLRs); b) Lectinas de unión a ácido siálico (siglecs); y c) galectinas [8–10]. Los CLRs comprenden una familia heterogénea de lectinas dependientes del catión Ca2+. Estas a su vez, se subdividen en dos categorías en base a la presencia de un motivo aminoacídico involucrado en el reconocimiento de glicanos y de la coordinación del Ca2+. La mayoría de estas lectinas poseen uno o más dominios de reconocimiento de carbohidratos (DRCs), y son expresadas en la superficie de un amplio espectro de células del sistema inmunológico, como macrófagos y células dendríticas (DCs). Los CLRs que contienen el motivo aminoacídico EPN (Glu-Pro-Asn), incluídos DC-SIGN, el receptor de manosa (MR) y tangerina, presentan afinidad por glicanos conteniendo manosa o fucosa. Por otro lado, CLRs conteniendo el motivo QPD (Gln-Pro-Asp) interactúan específicamente con N-acetilgalactosamina presente en posición terminal en glicanos. El receptor MGL (lectina de galactosa de macrófagos), presente en macrófagos y DCs, es un ejemplo de este grupo de lectinas [15]. Existen también CLRs que no poseen el requerimiento de Ca2+ para interactuar con glicanos, como el receptor Dectin-1 que reconoce específicamente β-glucanos de levaduras [16]. La interacción entre CLRs y glicanos específicos conduce a la internalización de los mismos por endocitosis. La señalización intracelular generada por endocitosis de CLRs promueve a su vez señales gatilladas por otros receptores importantes para el reconocimiento de peligro como los receptores de patrones asociados a patógenos de tipo Toll [17].
Las siglecs por su parte, poseen especificidad por glicanos que contienen ácido siálico. Algunas siglecs presentan un patrón de expresión restringido, mientras que otras se encuentran expresadas en varias células de linaje hematopoyético, tales como linfocitos B, macrófagos y eosinófilos. El reconocimiento de ácido siálico por parte de siglecs promueve el reclutamiento de fosfatasas de la familia SHP, las cuales atenúan vías de señalización que involucran fosforilación de residuos de tirosina [18]. Mientras que los CLRs y las siglecs actúan principalmente como proteínas transmembrana presentes en la superficie de células del sistema inmune, las galectinas son proteínas solubles secretadas al medio extracelular por una vía no canónica (sin intervención del sistema RE-Golgi). A nivel extracelular, las galectinas interactúan con una gran diversidad de receptores glicosilados por medio de interacciones proteína-glicanos y proteína-proteína, en un amplio espectro de tipos celulares [9, 10]. También se ha descripto que las galectinas pueden de manera intracelular, modular vías de señalización, regular la supervivencia de linfocitos y su actividad, e incluso interactuar con la maquinaria de splicing (corte y ensamblado) del ácido ribonucleico (ARN) [10, 19–23].
A la fecha se conocen 15 miembros de la familia de galectinas en mamíferos, aunque también se han encontrado proteínas de tipo galectinas en insectos, hongos y plantas [21, 24]. Las galectinas se subdividen en tres grupos en base a sus características estructurales, aunque todas presentan al menos un DRC de aproximadamente 130 aminoácidos conservado estructuralmente: las galectinas “prototipo” (galectina-1, -2, -5, -7, -10, -11, -13, -14 y -15) poseen un único DRC y pueden encontrarse como monómeros o como dímeros. Las galectinas de tipo “tándem” (galectina- 4, -6, -8, -9 y -12) contienen dos DRCs homólogos en tándem en una misma cadena polipeptídica, unidos por una región separadora (linker) de hasta 70 aminoácidos. Galectina-3 es la única referente del grupo “quimera”, la cual posee un único DRC con una región N-terminal de tipo no lectina, necesaria para su oligomerización (llegando a formar inclusive pentámeros) (Figura 2) [10, 20, 21, 23]. Inicialmente las galectinas fueron definidas por su habilidad de reconocer el disacárido N-acetil-lactosamina (Galβ(1-4)GlcNAc; LacNAc) presente en N- y O-glicanos en glicoproteínas de superficie [25]. Sin embargo estudios recientes revelaron que estas lectinas presentan diferencias sustanciales en las preferencias de glicanos, particularmente en el reconocimiento de estructuras sialiladas, sulfatadas o fucosiladas, como así también en la capacidad de reconocer LacNAc en posición terminal o en repeticiones internas en la estructura de los glicanos (Figura 2). Esta selectividad relativa podría representar, al menos en parte, la base molecular subyacente a las diferencias funcionales existentes entre miembros individuales de la familia [25–28].
La actividad biológica de las galectinas se encuentra influenciada por su estado de oligomerización, su estabilidad en microambientes reductores u oxidantes y la exposición de N- y O-glicanos en células blanco sobre las cuales ejercen su actividad [10]. Sumando complejidad al sistema, las galectinas también pueden reconocer glicanos presentes en microorganismos, lo cual sugiere un origen temprano para estas moléculas como proteínas solubles capaces de reconocer patrones glicosídicos asociados a patógenos [11, 29, 30]. Algunas galectinas presentan en mamíferos un patrón de expresión ubicuo mientras que otras exhiben una localización específica de tejidos. En el sistema inmunológico las galectinas se encuentran expresadas en prácticamente todas las células ya sea de manera constitutiva o inducible [10, 11, 20, 21, 23].
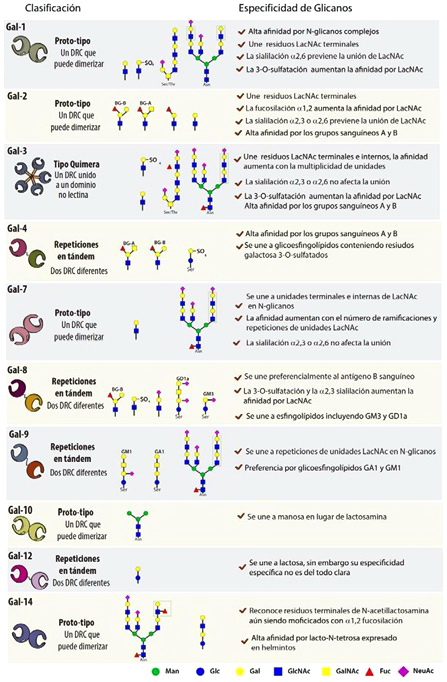
Figura 2: Clasificación de galectinas: estructura, función y especificidad sacarídica. En la figura se muestran la estructura y clasificación de las galectinas más estudiadas. Se observan representantes de los tres grupos de galectinas: galectinas ”proto-tipo” con un único DRC el cual puede homodimerizar (Gal-1, 2, 7, 10, 14); galectinas con repeticiones “en tándem” conteniendo dos DRCs diferentes unidos por una región de 70 aminoácidos (Gal-4, 8, 9, 12); y galectinas de tipo “quimera” con un DRC unido a una región no lectina que le permite oligomerizar, siendo Gal-3 su única representante. A su vez se muestran los ligandos preferidos de cada galectina y sus condiciones particulares de unión al mismo.
En esta revisión nos concentraremos en el papel de galectina-1 (Gal-1) como un regulador crucial de la homeostasis y tolerancia inmunológica, y un favor clave en el desarrollo de programas de vascularización. A su vez discutiremos las implicancias terapéuticas asociadas a procesos patológicos como inflamación crónica, autoinmunidad y cáncer, con especial énfasis en los aportes de nuestro equipo de investigación a la comprensión de estos procesos desde una perspectiva glicobiológica.
Gal-1: un nuevo mecanismo de escape tumoral y blanco terapéutico en cáncer
Nuestro sistema inmunológico cuenta con diversos tipos celulares (linfocitos NK, linfocitos CD8+ citotóxicos, linfocitos colaboradores o helper CD4+ TH1 y TH17, macrófagos, etc.) que poseen la capacidad potencial de reconocer células tumorales y eliminarlas con un alto grado de eficiencia. Sin embargo en la mayoría de los casos, ya sea por un estado generalizado de inmunosupresión o por el desarrollo de mecanismos específicos de inhibición a nivel local, el sistema inmunológico falla en la correcta eliminación de un tumor. Algunos tumores usurpan mecanismos de inmuno- privilegio o de resolución de la respuesta inmunológica desarrollando lo que comúnmente se denomina “mecanismo de escape tumoral”. Estos mecanismos se basan en moléculas inhibitorias capaces de suprimir la respuesta inmunológica, como son las vías gatilladas por PD-L1/PD-1 (del inglés Programmed cell death protein-1) y CTLA-4 (del inglés Cytotoxic T-Lymphocyte Antigen 4). Durante la resolución normal de un proceso inflamatorio (el montado para erradicar un microorganismo patógeno, para citar un ejemplo), estas moléculas se expresan en distintas células del sistema inmunológico, como por ejemplo los linfocitos T regulatorios (Tregs), lo cual permite el silenciamiento de la respuesta inmune, evitando así el daño a los tejidos cercanos causado por un exceso de inflamación. Los tumores por su parte, adquieren alguno de estos mecanismos, denominados puntos de control inmunológicos (o checkpoints), y los utilizan para su beneficio al inducir la muerte o parálisis de linfocitos que podrían eliminarlos.
De la comprensión de los mecanismos a través de los cuales los tumores escapan a la vigilancia inmunológica, surgieron en los últimos años distintas terapias que apuntan al bloqueo de estas vías de escape, mediante la administración de anticuerpos monoclonales bloqueantes de las moléculas inhibitorias CTLA-4 ó PD-1/PD-L1 [31–34]. Estos tratamientos apuntan a contrarrestar la inhibición que tumores ejercen sobre la respuesta inmunológica, permitiendo así que nuestro propio organismo logre erradicar al tumor. Estas terapias forman parte de un conjunto aún mayor denominado Inmunoterapia que involucra no sólo anticuerpos bloqueantes contra mecanismos de escape tumoral o moléculas co-inhibitorias, sino también anticuerpos agonistas de moléculas co-estimulatorias, vacunas de células tumorales o DCs o receptores quiméricos (CARs del inglés chimeric antigen receptors).
En un gran número de casos se han observado beneficios clínicos extraordinarios, incluyendo la reducción del tamaño tumoral y una mayor supervivencia de pacientes libres enfermedad en una gran variedad de tumores. Sin embargo, existen pacientes que son refractarios a estas terapias, e incluso hay algunos que responden inicialmente, pero luego de un tiempo sus tumores desarrollan mecanismos compensatorios, creciendo y diseminándose en diversos tejidos distantes (proceso conocido como metástasis). Estos mecanismos compensatorios incluyen, entre otros, a las moléculas inhibitorias llamadas LAG-3 y TIM-3, cuyas funciones autónomas pueden controlar diversos aspectos de la respuesta inmunológica antitumoral, promoviendo vías de escape alternativas [32–34].
Nuestro laboratorio ha estudiado durante varios años los mecanismos a través de los cuales Gal-1 es capaz de descifrar información relevante codificada por glicanos en receptores celulares del microambiente tumoral. En una primera aproximación, observamos que en muestras de pacientes con melanoma esta lectina se encontraba elevada en aquéllas lesiones con mayor potencial metastásico. Cuando la expresión de Gal-1 se silenciaba, en el modelo de melanoma B16 en ratones, el tumor era rechazado por su hospedador, siempre y cuando tuviera los repertorios de linfocitos T CD4 y CD8 intactos [35] (Figura 3). Descubrimos además que Gal-1 poseía la capacidad diferencial de inducir apoptosis de linfocitos T CD8, TH1 y TH17 (encargados en parte de la eliminación de tumores) a través de la interacción y segregación de los receptores CD45, CD43 y CD7 [36, 37], mientras que esto no ocurría con linfocitos TH2 o linfocitos T vírgenes [35,36] (Figura 3). Esta susceptibilidad diferencial se fundamentó en el hecho de que linfocitos T activados (CD8), CD4 TH1 y TH17 exhibían un “glicofenotipo” permisivo para la unión de Gal-1 (alta frecuencia de core-2-O-glicanos no sialilados), mientras que los linfocitos no susceptibles (TH2) poseían un escudo de ácido siálico en posición α2,6 que impedía la unión de Gal-1 a residuos de N-acetillactosamina terminales [10, 36]. De esta forma, los tumores expresan y secretan Gal-1 al medio extracelular, promoviendo el desarrollo de un microambiente anti-inflamatorio, el cual inhibe la respuesta inmunológica, ya sea a través de la eliminación de linfocitos anti-tumorales como también promoviendo circuitos tolerogénicos encargados de silenciar la respuesta inmune, como la expansión de linfocitos T regulatorios (TRegs) en el microambiente tumoral [38], entre otros (Figura 3). Tal es la importancia de Gal-1 en la evasión tumoral y posterior metástasis, que la expresión de esta lectina en ciertos tipos de tumores se acrecienta conforme el mismo se vuelve más maligno, de manera que la determinación de Gal-1 constituye un factor pronóstico clave en la progresión neoplásica [39].
En este contexto, en escenarios fisiológicos, Gal-1 participa en fenómenos de inmunosupresión claves que confieren privilegio inmunológico durante el embarazo. Hemos demostrado que la placenta produce altos tenores de Gal-1 en respuesta a la hormona progesterona generando una cascada jerárquica de inmunosupresión que permite el correcto desarrollo fetal durante el periodo de gestación [40–42].
Los resultados obtenidos por nuestro equipo, fueron reproducidos, en diversos modelos experimentales por distintos grupos de investigación, y nos condujeron a preguntarnos si Gal-1 podría resultar, al igual que PD-1 o CTLA-4, un punto de control inmunológico y un blanco de inmunoterapia. Comprobamos en diferentes modelos, incluidos el melanoma, cáncer de pulmón, cáncer de mama y sarcoma de Kaposi, que el bloqueo la acción de Gal-1 (ya sea por medio del silenciamiento génico o por la administración de un anticuerpo monoclonal neutralizante), disminuía el crecimiento tumoral, aumentaba la supervivencia de ratones tratados, disminuían su potencialidad metastático y confería sensibilidad a terapias anti-angiogénicas convencionales [38, 43]. Particularmente, comprobamos que Gal-1 constituía una vía de escape tumoral alternativa en tumores resistentes a la terapia anti–angiogénica bloqueante del factor de crecimiento endotelial vascular (VEGF), a través de su capacidad de promover el desarrollo de vasos sanguíneos (angiogénesis) mediante su interacción directa con N-glicanos complejos presentes en el receptor tipo 2 de VEGF (VEGFR2). Este mecanismo se gatilla en respuesta a la ausencia de oxígeno (hipoxia), fenómeno característico de ciertas regiones centrales del microambiente tumoral [43, 44] (Figura 3). De este modo, el tratamiento con un anticuerpo monoclonal anti-Gal-1 revirtió la resistencia de tumores refractarios a la terapia con anticuerpos bloqueantes anti-VEGF [43]. El tratamiento combinado con anticuerpos anti-VEGF/anti-Gal-1 logró a su vez normalizar la vasculatura permitiendo la correcta afluencia de linfocitos T CD8 capaces de reconocer y erradicar dichos tumores [43]. A su vez, recientemente observamos que la microbiota intestinal es capaz de promover tumorigénesis a distancia por medio de señales inflamatorias mediadas por interleuquina-6 (IL-6) y linfocitos Tγδ, y células mieloides supresoras productoras de Gal-1 [45]. Estas evidencias nos permitieron proponer a Gal-1 como un nuevo punto de chequeo inmunológico y un atractivo blanco terapéutico en diversos tipos de cáncer incluidos el cáncer de piel (melanoma), linfoma Hodgkin, leucemia linfática crónica (LLC), sarcoma de Kaposi, carcinoma de pulmón, mama, páncreas y próstata, glioblastoma y neuroblastoma [34, 46].
Gal-1: un potencial agente terapéutico para enfermedades autoinmunes e inflamatorias crónicas
Las propiedades inmunosupresoras y terapéuticas de Gal-1 se han evaluado en varios modelos de inflamación crónica y autoinmunidad, incluyendo encefalomielitis autoinmune experimental (EAE), artritis inducida por colágeno (AIC), colitis inducida por ácido 2,4,6-trinitrobenceno sulfónico (TNBS), uveítis autoinmune experimental, hepatitis inducida por concanavalina A y diabetes autoinmune experimental [47–53].
Inicialmente, estudiamos el papel que cumple Gal-1 en el desarrollo de artritis en modelos murinos. El modelo de artritis inducido por colágeno se logra por la inmunización con una agente que contiene colágeno tipo-II a los fines de generar una respuesta inmune, mediada por linfocitos T y linfocitos B [54]. La artritis reumatoidea (AR) se caracteriza por inflamación crónica en las articulaciones sinoviales, lo cual conduce a una hiperplasia en fibroblastos residentes y un aumento en el ingreso de linfocitos, macrófagos y células plasmáticas, las cuales presentan un perfil activado [55]. Estas células proliferan anormalmente, invadiendo tanto el cartílago como el hueso, produciendo un elevado número de citoquinas pro-inflamatorias, metaloproteasas y promoviendo la activación de osteoclastos [56]. Algunas consecuencias fisiopatológicas de la enfermedad pueden explicarse debido a una inadecuada apoptosis, la cual promueve la supervivencia de células T auto-reactivas y macrófagos. Empleando este modelo hemos podido demostrar el rol anti-inflamatorio de Gal-1 como supresor de las manifestaciones clínicas e histopatológicas de esta enfermedad. La administración diaria de Gal-1 recombinante humana en ratones con AIC o la administración de fibroblastos modificados genéticamente a los fines de expresar altos niveles de esta lectina, fueron capaces de suprimir las manifestaciones clínicas de la enfermedad, reducir los niveles de inmunoglobulinas (Ig)G anti-colágeno en suero de ratones tratados, y desviar la producción de citoquinas hacia un perfil tipo TH2, con disminución en los niveles de interferón-γ (IFN-γ) y aumento de IL-5 [49] (Figura 3).
Sumado a su efecto supresor sobre la respuesta inmune adaptativa, Gal-1 también demostró capacidad de regular la activación de células de la inmunidad innata. Los tratamientos con Gal-1 redujeron la infiltración de neutrófilos, la degranulación de mastocitos y la expresión en macrófagos de la óxido nítrico sintasa inducible (iNOS) [57, 58]. Sin embargo, cabe preguntarnos ¿Cuál es el panorama en pacientes con artritis? En pacientes pediátricos que padecían artritis idiopática juvenil (AIJ) se ha observado una disminución en la expresión de esta lectina en tejido sinovial [59]. Aunque los niveles plasmáticos de Gal-1 son comparables entre los pacientes adultos con AR y los controles sanos, la concentración de esta galectina en el líquido sinovial se encuentra significativamente disminuida [60]. Estos resultados cobran particular interés al proponer el diseño de fármacos similares a Gal-1 como posibles drogas en el tratamiento de la artritis reumatoidea y otras enfermedades autoinmunes.
Por otra parte Gal-1 demostró actividad inmunomodulatoria sobre la encefalomielitis autoinmune experimental (EAE), modelo animal que recapitula las manifestaciones clínicas, histopatológicas e inmunológicas de la esclerosis múltiple (EM). El protocolo de inducción de EAE también involucra el concepto de inmunización pero esta vez con extractos de proteínas provenientes de la materia blanca del sistema nervioso central (SNC), o antígenos peptídicos específicos [61]. Usualmente se utilizan: proteína básica de mielina (PBM), proteína proteolipídica (PLP) o glicoproteína oligodendrocitaria de mielina (MOG) [62]. La patología generada puede variar desde episodios de parálisis muscular aguda hasta procesos neurológicos recurrentes crónicos y discapacidad motora general [63]. Se conoce que las subpoblaciones de linfocitos TH1 y TH17 son las responsables de la severidad y persistencia del proceso autoinmune en la EM y la EAE [64, 65]. En función de la capacidad de Gal-1 de inhibir la proliferación y expansión de linfocitos T activados, mediante mecanismos que involucran bloqueo de la activación [66], arresto del ciclo celular [67] e inducción de apoptosis [68], estudiamos el impacto de esta lectina en la EAE. La inducción de enfermedad en animales deficientes en el gen de Gal-1 (Lgals1-/-) reveló un incremento en la severidad de la patología autoinmune desmielinizante. Estos ratones demostraron un extenso infiltrado inflamatorio y mayores áreas de desmielinización en secciones de médula espinal, un aumento en la frecuencia de linfocitos TH1 y TH17 en bazo, así como una mayor respuesta proliferativa antígeno-especifica. Estos resultados demostraron que Gal-1 endógena controla el desarrollo de procesos inflamatorios en el SNC, inhibiendo la severidad de procesos autoinmunes. Conforme a estos hallazgos, cuando ratones incluidos en un protocolo de EAE fueron tratados con Gal-1 recombinante, los mismos evidenciaron una disminución de la severidad clínica de la enfermedad y menor frecuencia y número absoluto de células TH17 y TH1 [36] (Figura 3).
Por otro lado, independientemente de su capacidad directa de modular la fisiología de células T, demostramos que Gal‐1 es capaz de generar DCs con un perfil tolerogénico. DCs expuestas a la acción de Gal‐1 fueron capaces de promover tolerancia de linfocitos T y resolver la inflamación autoinmune del SNC a través de un mecanismo dependiente de la secreción de IL‐27 e IL‐10. También observamos un incremento considerable de la expresión de Gal‐1 durante el pico y la fase de resolución de la EAE, tal como se evidenció para otras señales inhibitorias como PD‐L1 y su receptor PD‐1 [69] (Figura 3). Por último demostramos en este modelo, que Gal‐1 además de eliminar selectivamente linfocitos T patogénicos y promover circuitos tolerogénicos mediados por DCs, actúa directamente sobre la microglia del SNC, cambiando su patrón funcional de un perfil pro‐inflamatorio M1 hacia un perfil anti‐inflamatorio de tipo M2 [70] (Figura 3). Finalmente, estudios recientes de nuestro laboratorio revelaron un papel clave de Gal‐1 en la resolución de patologías alérgicas y asma a través del control de la capacidad migratoria y funcional de eosinófilos [71], ampliando las posibilidades inmunomodulatorias de esta molécula y sus ligandos. Estos hallazgos, en su conjunto, posicionan a Gal‐1 como un potencial agente terapéutico en el tratamiento de enfermedades autoinmunes y alérgicas con activación “exacerbada” del sistema inmunológico.
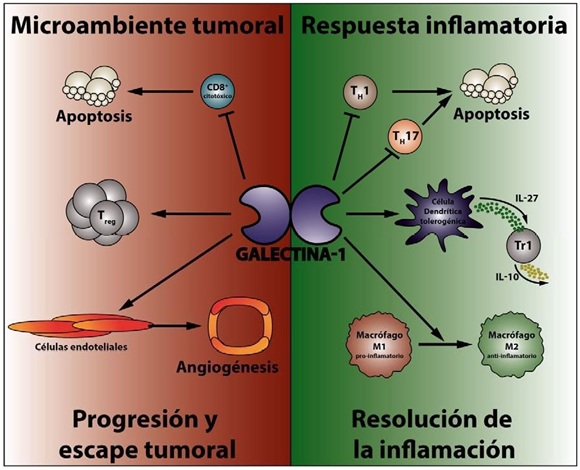
Figura 3: Gal-1 en autoinmunidad y cáncer: En este esquema se muestran de forma resumida las funciones de Gal-1 que fueron descriptas a lo largo de esta revisión. A la izquierda se observan todos los efectos deletéreos que ejerce Gal-1 sobre el sistema inmunológico en el microambiente tumoral, incluyendo la inducción de apoptosis de linfocitos T CD8+, la expansión de linfocitos TRegs inmunosupresores y la inducción de angiogénesis aberrante, fenómenos que favorecen la progresión tumoral. Por otro lado, a la derecha se esquematizan las funciones beneficiosas subyacentes a la administración de Gal-1 en enfermedades autoinmunes incluyendo la muerte celular de linfocitos TH1 y TH17, la diferenciación de células dendríticas (DCs) productoras de IL-27, las cuales perfilan la respuesta T a un fenotipo Tr1 productor de IL-10 y la polarización de macrófagos pro-inflamatorios M1 hacia un perfil anti-inflamatorio de tipo M2. De este modo, la administración de Gal-1 exógena favorecería la resolución de procesos inflamatorios y autoinmunes.
Conclusiones y Perspectivas
Gal-1, un miembro prototípico de la familia de galectinas con afinidad por N-acetillactosamina, ejerce un sinnúmero de funciones biológicas que impactan en diversos procesos fisiológicos y patológicos. En esta revisión hemos focalizado, a partir de los aportes realizados por nuestro equipo de investigación, en el papel crucial que desempeña Gal-1 en la homeostasis del sistema inmunológico en cáncer y enfermedades autoinmunes, y su protagonismo como factor pro- angiogénico no canónico en fenómenos de resistencia a terapias anti-angiogénicas convencionales.
En un contexto fisiológico, la eliminación de linfocitos T patogénicos y la activación de circuitos tolerogénicos mediado por Gal-1, resultaría indispensable para la correcta resolución de procesos inflamatorios y la generación de tolerancia inmunológica (Figura 3). Cuando los niveles de Gal-1 o su funcionalidad (ya sea por falta de actividad de la molécula per se, o por ausencia o enmascaramiento de los glicanos reconocidos por la misma) se encuentran alterados, estos pueden desencadenar distintas patologías:
- Por un lado, Gal-1 promueve escape tumoral favoreciendo la evasión del sistema inmunológico a través de la inducción de apoptosis de linfocitos T efectores, la expansión de células TREGs y la diferenciación de DCs hacia un perfil tolerogénico. Además, promueve la formación de nuevos vasos sanguíneos actuando en células endoteliales adyacentes al tumor, permitiendo el influjo de oxígeno y nutrientes. De este modo, en un contexto tumoral, Gal-1 se comportaría como un "villano" (Figura 3). Desde una perspectiva terapéutica, el bloqueo de Gal-1 (mediante un anticuerpo bloqueante por ejemplo) permitiría la supresión del crecimiento tumoral y la erradicación de metástasis a través de la potenciación de la respuesta inmune y la interrupción de circuitos angiogénicos.
- Por otro lado, en procesos inflamatorios descontrolados, al igual que en desórdenes autoinmunes como la AR, EM, uveítis, diabetes y enfermedad de Crohn, Gal-1 ejercería un efecto inmunosupresor selectivo sobre linfocitos T patogénicos TH1 y TH17 y promovería la activación de circuitos tolerogénicos, comportándose de este modo como una "heroína" (Figura 3). Desde una perspectiva terapéutica, administrar Gal-1 en procesos inflamatorios ayudaría a la resolución de dichos procesos, evitando o disminuyendo (dependiendo del grado de severidad de la patología) el posible daño a tejidos no involucrados.
Agradecimientos
Los autores agradecen el apoyo de la Agencia de Promoción Científica y Tecnológica (PICT V 2014-367; PICT 2012-2440), CONICET, Universidad de Buenos Aires y Fundaciones Sales y Bunge & Born. N.A.P., L.G.M. y S.M.M. agradecen su beca del CONICET.
Referencias:
1. Ohtsubo K, Marth JD (2006) Glycosylation in cellular mechanisms of health and disease Cell 126: 855–867 DOI: 10.1016/j.cell.2006.08.019.
2. Bard F, Chia J (2016) Cracking the Glycome Encoder: Signaling, Trafficking, and Glycosylation Trends in Cell Biology 26: 379–388 DOI: 10.1016/j.tcb.2015.12.004.
3. Moremen KW, Tiemeyer M, Nairn AV (2012) Vertebrate protein glycosylation: diversity, synthesis and function Nature Reviews Molecular Cell Biology. 13: 448–462 DOI: 10.1038/nrm3383.
4. Cerliani JP, Blidner AG, Toscano MA, Croci DO, Rabinovich GA (2016) Translating the “Sugar Code” into Immune and Vascular Signaling Programs. Trends in Biochemical Sciences. xx: 1–19 DOI: 10.1016/j.tibs.2016.11.003.
5. Marth JD, Grewal PK (2008) Mammalian glycosylation in immunity Nature Reviews Immunology. 8: 874– 887 DOI: 10.1038/nri2417.
6. Pinho SS, Reis CA (2015) Glycosylation in cancer: mechanisms and clinical implications Nature Reviews Cancer 15: 540–555 DOI: 10.1038/nrc3982.
7. Albrecht S, Unwin L, Muniyappa M, Rudd PM (2014) Glycosylation as a marker for inflammatory arthritis Cancer Biomarkers 14: 17–28 DOI: 10.3233/CBM-130373.
8. Johnson JL, Jones MB, Ryan SO, Cobb BA (2013) The regulatory power of glycans and their binding partners in immunity Trends in Immunology 34: 290–298 DOI: 10.1016/j.it.2013.01.006.
9. Rabinovich GA, Croci DO (2012) Regulatory Circuits Mediated by Lectin-Glycan Interactions in Autoimmunity and Cancer Immunity 36: 322–335 DOI: 10.1016/j.immuni.2012.03.004.
10. Rabinovich GA, Toscano MA (2009) Turning “sweet” on immunity: galectin-glycan interactions in immune tolerance and inflammation Nature Reviews Immunology 9: 338–352 DOI: 10.1038/nri2536.
11. van Kooyk Y, Rabinovich GA (2008) Protein-glycan interactions in the control of innate and adaptive immune responses Nature Immunology. 9: 593–601 DOI: 10.1038/ni.f.203.
12. Dam TK, Brewer CF (2010) Maintenance of cell surface glycan density by lectin-glycan interactions: a homeostatic and innate immune regulatory mechanism Glycobiology 20: 1061–1064 DOI: 10.1093/glycob/cwq084.
13. Dennis JW, Nabi IR, Demetriou M (2009) Metabolism, cell surface organization, and disease Cell 139: 1229–1241 DOI: 10.1016/j.cell.2009.12.008.
14. Nabi IR, Shankar J, Dennis JW (2015) The galectin lattice at a glance Journal of Cell Science 128: 2213 2219 DOI: 10.1242/jcs.151159.
15. Drickamer K (1999) C-type lectin-like domains Current Opinions in Structure Biology 9: 585–590.
16. Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, et al (2005) Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins Immunity 22: 507–517 DOI: 10.1016/j.immuni.2005.03.004.
17. Osorio F, Reis e Sousa C (2011) Myeloid C-type lectin receptors in pathogen recognition and host defense Immunity 34: 651–664 DOI: 10.1016/j.immuni.2011.05.001.
18. Crocker PR, Paulson JC, Varki A (2007) Siglecs and their roles in the immune system Nature Reviews Immunol. 7: 255–266 DOI: 10.1038/nri2056.
19. Liu F-T, Patterson RJ, Wang JL (2002) Intracellular functions of galectins Biochimica et Biophysica Acta 1572: 263–273.
20. Rabinovich GA, Toscano MA, Jackson SS, Vasta GR (2007) Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 17: 513–520 DOI: 10.1016/j.sbi.2007.09.002. [21] Yang R-Y, 21.
21. Rabinovich GA, Liu F-T (2008) Galectins: structure, function and therapeutic potential Expert Reviews in Molecular Medicine 10: e17 DOI: 10.1017/S1462399408000719.
22. Kubach J, Lutter P, Bopp T, Stoll S, Becker C, Huter E, et al (2007) Human CD4+CD25+ regulatory T cells: proteome analysis identifies galectin-10 as a novel marker essential for their anergy and suppressive function Blood 110: 1550–1558 DOI: 10.1182/blood-2007-01-069229.
23. Cummings RD, Liu F-T (2009) Galectins. In Essentials of Glycobiology., 2° Edition. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart, and M. E. Etzler, eds. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY).
24. Vasta GR (2012) Galectins as pattern recognition receptors: structure, function, and evolution Advances in Experimental Medicine and Biology 946: 21–36 DOI: 10.1007/978-1-4614-0106-3_2.
25. Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, et al (2002) Oligosaccharide specificity of galectins: a search by frontal affinity chromatography Biochimca et Biophysica Acta 1572: 232–254.
26. Patnaik SK, Potvin B, Carlsson S, Sturm D, Leffler H, Stanley P (2006) Complex N-glycans are the major ligands for galectin-1, -3, and -8 on Chinese hamster ovary cells Glycobiology 16: 305–317 DOI: 10.1093/glycob/cwj063.
27. Stowell SR, Arthur CM, Mehta P, Slanina KA, Blixt O, Leffler H, et al (2008) Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens Journal of Biological Chemistry 283: 10109–10123 DOI: 10.1074/jbc.M709545200.
28. Ideo H, Matsuzaka T, Nonaka T, Seko A, Yamashita K (2011) Galectin-8-N-domain recognition mechanism for sialylated and sulfated glycans Journal of Biological Chemistry. 286: 11346–11355 DOI: 10.1074/jbc.M110.195925.
29. Rabinovich GA, Gruppi A (2005) Galectins as immunoregulators during infectious processes: from microbial invasion to the resolution of the disease Parasite Immunology. 27: 103–114 DOI: 10.1111/j.1365-3024.2005.00749.x.
30. Vasta GR (2009) Roles of galectins in infection Nature Reviews Microbiology 7: 424–438 DOI: 10.1038/nrmicro2146.
31. Topalian SL, Drake CG, Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy Cancer Cell 27: 450–461 DOI: 10.1016/j.ccell.2015.03.001.
32. Sharma P, Allison JP (2015) Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential Cell 161: 205–214 DOI: 10.1016/j.cell.2015.03.030.
33. Anderson AC, Joller N, Kuchroo VK. (2016) Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 44: 989–1004 DOI: 10.1016/j.immuni.2016.05.001.
34. Méndez-Huergo SP, Blidner AG, Rabinovich GA (2017) Galectins: emerging regulatory checkpoints linking tumor immunity and angiogenesis Current Opinion in Immunology. 45: 8–15 DOI: 10.1016/j.coi.2016.12.003.
35. Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, et al (2004) Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell- mediated rejection; A potential mechanism of tumor-immune privilege Cancer Cell 5: 241–251.
36. Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, et al (2007) Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death Nature Immunology 8: 825–834 DOI: 10.1038/ni1482.
37. Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu F-T, et al (2006) Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. Journal of Immunology 176: 778–789.
38. Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon SL, Mendez- Huergo SP, et al (2013) Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease Cancer Research 73: 1107–1117 DOI: 10.1158/0008-5472.CAN-12-2418.
39. Laderach DJ, Gentilini LD, Giribaldi L, Delgado VC, Nugnes L, Croci DO, et al (2013) A unique galectin signature in human prostate cancer progression suggests galectin-1 as a key target for treatment of advanced disease. Cancer Research 73: 86–96 DOI: 10.1158/0008-5472.CAN-12-1260.
40. Blois SM, Ilarregui JM, Tometten M, Garcia M, Orsal AS, Cordo-Russo R, et al (2007) A pivotal role for galectin-1 in fetomaternal tolerance Nature Medicine 13: 1450–1457 DOI: 10.1038/nm1680.
41. Ramhorst RE, Giribaldi L, Fraccaroli L, Toscano MA, Stupirski JC, Romero MD, et al (2012) Galectin-1 confers immune privilege to human trophoblast: implications in recurrent fetal loss Glycobiology 22: 1374–1386 DOI: 10.1093/glycob/cws104.
42. Blidner AG, Rabinovich GA (2013) “Sweetening” pregnancy: galectins at the fetomaternal interface American Journal of Reproductive Immunology. 69: 369–382 DOI: 10.1111/aji.12090.
43. Croci DO, Cerliani JP, Dalotto-Moreno T, Mendez-Huergo SP, Mascanfroni ID, Dergan-Dylon SL, et al (2014) Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156: 744–758 DOI: 10.1016/j.cell.2014.01.043.
44. Croci DO, Salatino M, Rubinstein N, Cerliani JP, Cavallin LE, Leung HJ, et al (2012) Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. Journal of Experimental Medicine. 209: 1985–2000 DOI: 10.1084/jem.20111665.
45. Rutkowski MR, Stephen TL, Svoronos N, Allegrezza MJ, Tesone AJ, Perales-Puchalt A, et al (2015) Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation Cancer Cell 27: 27–40 DOI: 10.1016/j.ccell.2014.11.009.
46. Rabinovich GA, Conejo-Garcia JR (2016) Shaping the Immune Landscape in Cancer by Galectin-Driven Regulatory Pathways Journal of Molecular Biology 428: 3266–3281 DOI: 10.1016/j.jmb.2016.03.021.
47. Ilarregui JM, Bianco GA, Toscano MA, Rabinovich GA (2005) The coming of age of galectins as immunomodulatory agents: impact of these carbohydrate binding proteins in T cell physiology and chronic inflammatory disorders Annals of the Rheumatic Diseases 64 Suppl 4: iv96-103 DOI: 10.1136/ard.2005.044347.
48. Mari ER, Rasouli J, Ciric B, Moore JN, Conejo-Garcia JR, Rajasagi N, et al (2016) Galectin-1 is essential for the induction of MOG35-55 -based intravenous tolerance in experimental autoimmune encephalomyelitis European Journal of Immunology 46: 1783–1796 DOI: 10.1002/eji.201546212.
49. Rabinovich GA, Daly G, Dreja H, Tailor H, Riera CM, Hirabayashi J, et al (1999) Recombinant galectin-1 and its genetic delivery suppress collagen-induced arthritis via T cell apoptosis Journal of Experimental Medicine 190: 385–398.
50. Santucci L, Fiorucci S, Rubinstein N, Mencarelli A, Palazzetti B, Federici B, et al (2003) Galectin-1 suppresses experimental colitis in mice Gastroenterology 124: 1381–1394.
51. Toscano MA, Commodaro AG, Ilarregui JM, Bianco GA, Liberman A, Serra HM, et al (2006) Galectin-1 suppresses autoimmune retinal disease by promoting concomitant Th2- and T regulatory-mediated anti-inflammatory responses Journal of Immunology 176: 6323–6332.
52. Santucci L, Fiorucci S, Cammilleri F, Servillo G, Federici B, Morelli A (2000) Galectin-1 exerts immunomodulatory and protective effects on concanavalin A-induced hepatitis in mice Hepatology 31: 399–406 DOI: 10.1002/hep.510310220.
53. Perone MJ, Larregina AT, Shufesky WJ, Papworth GD, Sullivan MLG, Zahorchak AF, et al (2006) Transgenic galectin-1 induces maturation of dendritic cells that elicit contrasting responses in naive and activated T cells Journal of Immunology. 176: 7207–7220.
54. Rabinovich GA (2000) Apoptosis as a target for gene therapy in rheumatoid arthritis Memórias do Instituto Oswaldo Cruz 95 Suppl 1: 225–233.
55. Feldmann M, Brennan FM, Maini RN (1996) Rheumatoid arthritis. Cell 85: 307–310.
56. Takayanagi H, Juji T, Miyazaki T, Iizuka H, Takahashi T, Isshiki M, et al (1999) Suppression of arthritic bone destruction by adenovirus-mediated csk gene transfer to synoviocytes and osteoclasts. Journal of Clinical Investigation 104: 137–146 DOI: 10.1172/JCI6093.
57. Rabinovich GA, Sotomayor CE, Riera CM, Bianco I, Correa SG (2000) Evidence of a role for galectin-1 in acute inflammation. European Journal of Immunology. 30: 1331–1339 DOI: 10.1002/(SICI)1521- 4141(200005)30:5<1331::AID-IMMU1331>3.0.CO;2-H.
58. Correa SG, Sotomayor CE, Aoki MP, Maldonado CA, Rabinovich GA (2003) Opposite effects of galectin-1 on alternative metabolic pathways of L-arginine in resident, inflammatory, and activated macrophages Glycobiology 13: 119–128 DOI: 10.1093/glycob/cwg010.
59. Harjacek M, Diaz-Cano S, De Miguel M, Wolfe H, Maldonado CA, Rabinovich GA (2001) Expression of galectins-1 and -3 correlates with defective mononuclear cell apoptosis in patients with juvenile idiopathic arthritis Journal of Rheumatology 28: 1914–1922.
60. Xibille-Friedmann D, Bustos Rivera-Bahena C, Rojas-Serrano J, Burgos-Vargas R, Montiel-Hernandez JL (2013) A decrease in galectin-1 (Gal-1) levels correlates with an increase in anti-Gal-1 antibodies at the synovial level in patients with rheumatoid arthritis Scandinavian Journal of Rheumatology 42: 102–107 DOI: 10.3109/03009742.2012.725769.
61. Baxter AG (2007) The origin and application of experimental autoimmune encephalomyelitis Nature Reviews Immunology 7: 904–912 DOI: 10.1038/nri2190.
62. Steinman L (1999) Assessment of animal models for MS and demyelinating disease in the design of rational therapy Neuron 24: 511–514.
63. Furlan R, Cuomo C, Martino G (2009) Animal models of multiple sclerosis Methods in Molecular Biology. 549: 157–173 DOI: 10.1007/978-1-60327-931-4_11.
64. Steinman L (1996) Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 85: 299–302.
65. Bettelli E, Oukka M, Kuchroo VK (2007) T(H)-17 cells in the circle of immunity and autoimmunity Nature Immunology 8: 345–350 DOI: 10.1038/ni0407-345.
66. Chung CD, Patel VP, Moran M, Lewis LA, Miceli MC (2000) Galectin-1 induces partial TCR zeta-chain phosphorylation and antagonizes processive TCR signal transduction Journal of Immunology 165: 3722–3729.
67. Blaser C, Kaufmann M, Muller C, Zimmermann C, Wells V, Mallucci L, et al (1998) Beta- galactoside-binding protein secreted by activated T cells inhibits antigen-induced proliferation of T cells European Journal of Immunology 28: 2311–2319.
68. Perillo NL, Pace KE, Seilhamer JJ, Baum LG (1995) Apoptosis of T cells mediated by galectin-1 Nature 378: 736–739 DOI: 10.1038/378736a0.
69. Ilarregui JM, Croci DO, Bianco GA, Toscano MA, Salatino M, Vermeulen ME, et al (2009) Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10 Nature Immunology 10: 981–991 DOI: 10.1038/ni.1772.
70. Starossom SC, Mascanfroni ID, Imitola J, Cao L, Raddassi K, Hernandez SF, et al (2012) Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration Immunity 37: 249–263 DOI: 10.1016/j.immuni.2012.05.023.
71. Ge XN, Ha SG, Greenberg YG, Rao A, Bastan I, Blidner AG, et al (2016) Regulation of eosinophilia and allergic airway inflammation by the glycan-binding protein galectin-1 Proceedings of the National Academy of Science of the United States of America 113: E4837-46 DOI: 10.1073/pnas.1601958113.
G.A. Ravinovich es profesor titular e investigador superior de CONICET, N.A. Pinto, L.G. Morosi y S. M. Maller son becarios doctorales de CONICET
|
Revista QuímicaViva Número 1, año 16, Abril 2017 quimicaviva@qb.fcen.uba.ar |