Nanomedicinas: lo esencial es invisible…al microscopio óptico
Eder Lilia Romero y María José Morilla.
Programa de Nanomedicinas, Departamento de Ciencia y Tecnologia, Universidad Nacional de Quilmes.
Recibido 1 de octubre de 2013- Aceptado 29 de Octubre de 2013
Resumen
El poder de resolución del microscopio óptico es de unos 200 nm y si
tenemos suerte, los nano-objetos más voluminosos apenas se distinguirán
como puntos fluorescentes. ¿Podremos, mediante el control de la
arquitectura de esos “puntos brillantes”, dirigir su acceso a ciertos
sectores del cuerpo, donde ejecutarían exitosamente funciones
terapéuticas? Por ahora estamos más lejos de lo que creemos en
materializar ese ejercicio mental. Sin embargo, de todas las
aplicaciones de la Nanotecnología al área de la salud, el campo
terapéutico es el más importante. En este artículo nos adentraremos en
los fenómenos que explican cómo funcionan los nano-objetos que
transportan principios activos hasta los sitios blancos. A partir de
allí será posible construir expectativas razonables sobre la irrupción
de esta nueva tecnología en el corto futuro. Sus potencialidades y
limitaciones son dependientes de nuestra capacidad de diseño racional,
de desarrollo industrial y conocimiento de los fenómenos de
nanotoxicidad, respectivamente.
Palabras clave: nano-objeto, efecto de incremento de permeación y
retención (EPR), nanomedicinas antitumorales
Nanomedicines, the essential is invisible…
to the light microscope
Abstract
The resolution power of the optical microscope is nearly 200 nm and if
we are lucky, the biggest nano-objects are saw as highly fluorescent
dots. How the architecture of these shiny dots be controlled, to target
them to specific body sites and successfully exert therapeutic actions?
For the moment, we have been unable of meeting such issue. Nonetheless,
drug delivery is by far the most important field of all the applications
of Nanotechnology in health. In this review, we will address the
phenomena underlying the action of nano-objects as drug delivery
systems. On that knowledge, it will be possible to build reasonable
expectancies to the irruption of these new medicines, in the near
future. Potentialities and pitfalls of nanomedicines will depend on our
skills for rational design, industrial development and insights gained
on the new phenomena of nanotoxicity.
Key words: Nano-objects, Enhanced Permeation and retention (EPR) effect,
antitumoral nanomedicines
1. Introducción
La Nanotecnología irrumpió en la comunidad científica hace más de 50
años [1], como una promesa capaz de resolver retos ingenieriles
inabordables para la materia macro o microestructurada. El propósito de
esta tecnología es diseñar nano-objetos, estudiar sus propiedades
estructurales y luego aplicarlos con múltiples fines. Los nano-objetos
se definen como objetos con una (nanoplato), dos (nanotubo) o tres
(nanopartícula) dimensiones en la nano-escala de tamaño. La nano-escala
posee un límite inferior de 1 nm (excluye a los átomos) y un límite
superior difuso que supera los 100 nm cuando los nuevos fenómenos
asociados al tamaño del nano-objeto no son de naturaleza cuántica, sino
por ejemplo, relativos a sus modalidades de captura y tráfico
intracelular. Nanopartículas de idénticas dimensiones, pueden presentar
distintas propiedades de acuerdo al material con que se las prepare. Por
otro lado, moléculas producidas por síntesis química o biológica y cuya
actividad dependa de su estructura primaria (secuencia de átomos o de
moléculas), no son considerados nano-objetos. Para bien o mal, desde
hace unos 10 años usamos diariamente productos que contienen
nano-objetos o superficies nanoestructuradas, a veces sin enterarnos.
Otras, la intervención de la Nanotecnología en campos como el de la
salud suscita tanto en legos como en la mayoría de los académicos, una
expectativa cercana a la idealización. Es que la idea de emplear
nano-objetos para resolver exitosamente complejos problemas de salud
resulta fascinante. La disciplina en cuestión se conoce como
Nanomedicina y emplea nano-objetos diseñados ad-hoc para operar en la
interfase entre el tamaño atómico y el celular con fines terapéuticos,
profilácticos y de tecnología médica (que incluye ingeniería de tejidos
y diagnostico).
No es posible identificar un único tipo de nano-objeto igualmente
útil para todos los propósitos. Por lo general, aquellos que se emplean
in vitro o ex vivo (como en la mayoría de las técnicas diagnósticas) no
siempre pueden administrarse a un ser vivo. Asimismo, en terapéutica y
profilaxis los nano-objetos son biodegradables y son aplicados por
distintas rutas. La mayoría de las estrategias terapéuticas están
basadas en la incorporación de un principio activo (PA) a un determinado
nano-objeto. A partir de allí, la farmacocinética, biodistribución y
tráfico intracelular del PA se independiza de su estructura química y
pasa a depender de la naturaleza estructural del nano-objeto. Por
volumen de ventas y patentes, la aplicación más importante de la
Nanotecnología en salud es la pertinente al campo terapéutico conocida
como nanotecnología farmacéutica [2]. Dicho esto, en este artículo
comentaremos los fenómenos que ocurren cuando los nano-objetos
(nanopartículas) se administran con fines terapéuticos por ruta
endovenosa. A diferencia de la inhalatoria, los fenómenos asociados a la
ruta endovenosa están bastante bien conocidos, además de poner en juego
la máxima capacidad operativa de los nano-objetos. Por su complejidad,
excluiremos referirnos al cruce de la barrera hematoencefalica.
Asimismo, no abordaremos la exposición no intencional de nano-objetos no
biodegradables, que tiene lugar fundamentalmente por ruta inhalatoria.
2. ¿Cómo funcionan las nanomedicinas?
2.1 El turbulento viaje de los nano-objetos
Una vez inyectados endovenosamente, los nano-objetos ingresan
directamente al corazón, desde cuyo ventrículo derecho -en humanos- son
bombeadas a los pulmones a un flujo de 5,6 litros por minuto [3]. Su
viaje por sangre venosa continúa hacia los capilares alveolares
pulmonares donde se vuelcan a la sangre arterial. Desde allí retornan al
corazón, para ser bombeadas en la sangre arterial a cada uno de los
órganos corporales. La magnitud de su acceso a los órganos dependerá del
volumen de flujo que accede a cada uno. Asimismo durante casi toda su
trayectoria, los nano-objetos permanecen confinados al compartimiento
vascular. Nano-objetos en el orden de los 100 nm de diámetro (unas 10
veces mayor que el diámetro hidrodinámico de una proteína plasmática
como la albúmina, aunque unas 100 veces menor que un glóbulo rojo) no
pueden difundir a través del endotelio vascular/capa de músculo liso
/membrana basal, ni penetrar los poros de la vasculatura (<6 nm en
vénulas post capilares). Más aun, los nano-objetos que pierdan su
estabilidad coloidal en circulación y se agreguen, pueden ocluir redes
vasculares particularmente angostas, como los capilares pulmonares
(entre 2 y 23 mm de diámetro). Al igual que las proteínas plasmáticas,
nano-objetos lo suficientemente pequeños pueden atravesar libremente las
fenestraciones de entre 100 - 150 nm (alrededor de 107 nm en humano) de
los sinusoides hepáticos (vasos de 5 -10 μm diámetro sin membrana basal
para acceder al espacio perisinusoidal o espacio de Disse), donde se
ubican los cordones de hepatocitos [4, 5]. Pero a diferencia de las
proteínas, los nano-objetos son reconocidos como extraños y fagocitados
por macrófagos fijos ubicados estratégicamente en la luz de los
sinusoides (células de Kupffer). Por su gran volumen, el hígado es el
órgano corporal con mayor capacidad de captura global de nano-objetos,
aunque el bazo lo supere en tasa de captura por unidad de masa de
tejido. Los órganos con vasculatura fenestrada como el hígado, bazo (con
anchos sinusoides (5-8 µm) en la vasculatura de pulpa roja, para dejar
pasar glóbulos rojos senescentes) y en menor medida, medula ósea, son
los únicos donde los nano-objetos pueden extravasar hacia los
intersticios tisulares. Claramente es la arquitectura vascular la
responsable directa de la biodistribución de nano-objetos inyectados
endovenosamente.
Luego de inyectados, además de diluirse y ser arrastrados en el
turbulento flujo vascular, los nano-objetos son inmediatamente cubiertos
por proteínas plasmáticas. Primeramente ocurre una cobertura
inespecífica y transitoria con albúmina, la proteína de mayor
concentración (35-40 g/l), que es rápidamente reemplazada por proteínas
de menor concentración pero mayor afinidad. Estas últimas (fibronectina,
fibrinógeno, proteína C reactiva, inmunoglobulinas, proteínas del
complemento C3b y C4b y lipoproteínas entre otras) se conocen como
opsoninas, o proteínas plasmáticas para las que los macrófagos expresan
receptores en su superficie. Las opsoninas permiten establecer múltiples
puntos de contacto entre la superficie de microorganismos o nano-objetos
y receptores fagociticos como los de la fracción Fc de anticuerpos
plasmáticos (R-Fc), de fosfatidilserina (R-PS) y de ciertas fracciones
del complemento (R-C) en la membrana de macrófagos [6, 7]. Una vez
opsonizados tal como si fueran microorganismos, la membrana plasmática
del macrófago se extiende alrededor de los nano-objetos, encerrándolos.
La vesícula así formada se conoce como fagosoma. El material dentro del
fagosoma es procesado en una ruta destructiva, que recibe los contenidos
de endosomas y lisosomas y culmina con la eliminación de sus fragmentos
hidrolizados. Recientemente ha comenzado a establecerse la lógica entre
el patrón y extensión de la opsonización -y por ende de la fagocitosis-
con el tamaño, geometría y naturaleza superficial del nano-objeto.
El bajo radio de curvatura de grandes nano-objetos (> 200 nm)
facilita la deposición de opsoninas y su posterior agregación (pérdida
de estabilidad coloidal) [8]. En general, es la pérdida de estabilidad
coloidal la responsable de la captura fagocítica de los nano-objetos [9,
10]. Los nano-objetos de superficie hidrofóbica atraen mayor proporción
de lipoproteínas y apolipoproteinas. Las lipoproteínas pueden
intercambiar lípidos con liposomas y nanopartículas lipídicas y
desestabilizar su estructura [11]. Una interacción de este tipo puede
ser responsable del targeting de nano-objetos a células que tengan
receptores para apolipoproteínas, como hepatocitos (con receptores para
apo E) [12] y sistema nervioso central (con receptores para apo A1, Apo
B100, apo E) [13]. En general las superficies negativa o positivamente
cargadas, muy hidrofóbicas o irregulares también promoverán la adhesión
de proteínas del complemento
Si bien es prácticamente imposible evitar la opsonización, la
agregación puede minimizarse modificando la superficie de los
nano-objetos. Esto se logra cubriéndolos con un polímero hidrofílico de
conformación adecuada, de unas decenas de angstroms de espesor. Para tal
fin usualmente se emplea polietilenglicol de peso molecular 2000
(PEG2000), y la estrategia que se conoce como “estabilización estérica”
[14]. La estabilización estérica mediante PEG, tiene un rol
controvertido ya que además puede ser responsable de toxicidad, como
veremos más adelante.
Los nano-objetos estabilizados estéricamente pueden permanecer en
circulación por largo tiempo sin ser fagocitados. La evasión temporal de
la fagocitosis es el primer paso para que los nano-objetos puedan
acceder a tejidos diferentes al hígado, bazo o medula ósea.
2.2 Incremento local de permeación y retención: el efecto EPR
La estabilización estérica de los nano-objetos aumenta sus
posibilidades de extravasar en sitios con elevada permeabilidad
vascular. Tales sitios están presentes en ciertas patologías, como los
tumores sólidos (que constituyen cerca del 85% de los cánceres humanos).
En la tabla 1, se muestran ejemplos de diferentes nanomedicinas
antitumorales en uso clínico y en ensayos clínicos avanzados. El
delivery de nano-objetos desde el sitio de inyección hasta las células
tumorales involucra su transporte por circulación sistémica,
extravasación y transporte a través del intersticio tumoral.
A partir de un tamaño mínimo, los tumores sólidos comienzan a
estimular su propia vascularización [15]. La resultante vasculatura
anómala asociada al drenaje linfático defectuoso, son responsables de la
acumulación y retención sitio especifica de nano-objetos [16]. Este
fenómeno observado en ciertos tumores sólidos, sobre todo aquellos con
elevado grado de malignidad, es conocido como efecto de incremento de
permeabilidad y retención (enhanced permeability and retention (EPR)
effect) o efecto EPR [17, 18].
2.2. i Factores locales responsables del incremento de la permeabilidad
vascular
Los tumores sobre-expresan múltiples isoformas del factor de
crecimiento endotelial vascular VEGF-idéntico al factor de permeabilidad
vascular (VPF) [19, 20]. Los VEGF pertenecen a una familia de mitogenos
endoteliales y mediadores de permeabilidad vascular, con diferentes
capacidades para interactuar con proteínas de la matriz extracelular
[21]. Como resultado, la vasculatura tumoral es heterogénea y no sigue
una morfología estándar (arteriolas, capilares y vénulas en una red
organizada, de ramificación dicótoma con orden jerárquico) [22]. La
vasculatura tumoral es dilatada, sacular y mal alineada, con células
endoteliales defectivas y amplias fenestraciones entre 300 y 4700 nm,
ausencia de musculo liso o inervación, amplio lumen y sin receptores
funcionales para angiotensina II [23-25] (Figura 1). En ella la
dirección del flujo sanguíneo puede cambiar abruptamente, cesar o ser
errático [26].
Desde hace unos 30 años se sabe que la permeabilidad vascular propia
de inflamaciones e inducida por infecciones es gatillada por
bradiquinina (BK, quinina), vía activación de una cascada proteolítica
[27]. Luego se descubrió que la BK también acompaña procesos
carcinomatosos, siendo responsable de la acumulación de fluido pleural y
ascítico y de dolor [28]. Además, los tumores sólidos expresan
simultáneamente múltiples mediadores inflamatorios como el óxido nítrico
(NO) (un mediador de vasodilatación, hipotensión, angiogénesis,
proliferación celular y extravasación); prostaglandinas (PGs),
sintetizadas por ciclo-oxigenasas (COX)-1 y-2 y peroxinitrito (ONOO-),
formado por una rápida reacción entre los radicales superóxido (O2-) y
NO y activador de pro-matriz metalproteinasas (MMPs), estas últimas
enzimas que degradan la matriz extracelular [29, 30].
Tabla 1. Ejemplos de diferentes nanomedicinas antitumorales en uso
clínico y en ensayos clínicos avanzados
|
Compuesto |
Nombre |
Indicación |
Estatus (año) |
|
Doxorubicina liposomal peguilada |
Myocet, Caelyx (Doxil) |
Cáncer de ovario y mama, refractarios, Sarcoma de Kaposi asociado a HIV, mieloma múltiple |
Aprobado FDA (1995) |
|
Daunorubicina liposomal |
Daunoxome |
Sarcoma de Kaposi asociado a HIV |
Aprobado (1996) |
|
Doxorubicina liposomal no pequilado |
Myocet |
Cáncer de mama metastásico |
Aprobado en Europa y Canada (año) |
|
Vincristina liposomal |
Marqibo |
Leucemia linfoblástica aguda con cromosoma philadelphia negativo (Ph-) |
Aprobado (2012) |
|
Doxorubicina liposomas termosensibles |
ThermoDox |
Carcinoma hepatocelular |
Fase III |
|
Cisplatino liposomal |
Lipoplatin |
Carcinoma de células no pequeñas de pulmón y pancreático |
Fase II y III |
|
Lurtotecan liposomal |
OSI-221 |
Varios |
Fase II |
|
Paclitaxel en nanopartículas de albúmina |
Abraxane |
Cáncer de mama |
Aprobado (2005) |
|
L-asparginasa-Peg |
Oncaspar |
Leucemia aguda linfocítica |
Aprobado (1994) |
|
Paclitaxel-Acido poliglutamico |
Opaxio |
Carcinoma de células no pequeñas de pulmón, de ovario y otros |
Fase III |
|
Doxetacel en nanopartículas de albúmina |
ABI-008 |
Cáncer de próstata refractario a hormonas |
Fase II |
|
Paclitaxel-micelas poliméricas |
NK105 |
Cáncer de estómago |
Fase III |
|
Doxorubicina- micelas poliméricas |
NK911 |
Varios |
Fase II completa |
|
Cisplatino- micelas poliméricas |
Nanoplatin |
Varios |
Fase I |
Adaptado de Duncan R, Gaspar R (2011)
Nanomedicine(s) under the microscope. Molecular Pharmaceutics 8:
2101-2141
2.2. ii Disminución del drenaje linfático
La red de linfáticos remueve macromoléculas y fluido intersticial y
es esencial para las funciones inmunes del tejido normal y mantenimiento
del balance de fluidos en el intersticio. En los tumores sólidos sin
embargo, el drenaje linfático es usualmente defectivo [15, 31]. Los
vasos linfáticos son comprimidos por las propias células tumorales [32]
y por ende su funcionalidad depende de su localización. Los linfáticos
periféricos o en la interfase tumoral son funcionales, no así los del
interior tumoral [33]. La funcionalidad reducida de los linfáticos está
asociada a flujo retrógrado, con invasión de células tumorales a vasos
linfáticos periféricos y promoción de metástasis dentro del sistema
linfático [26, 34, 35].
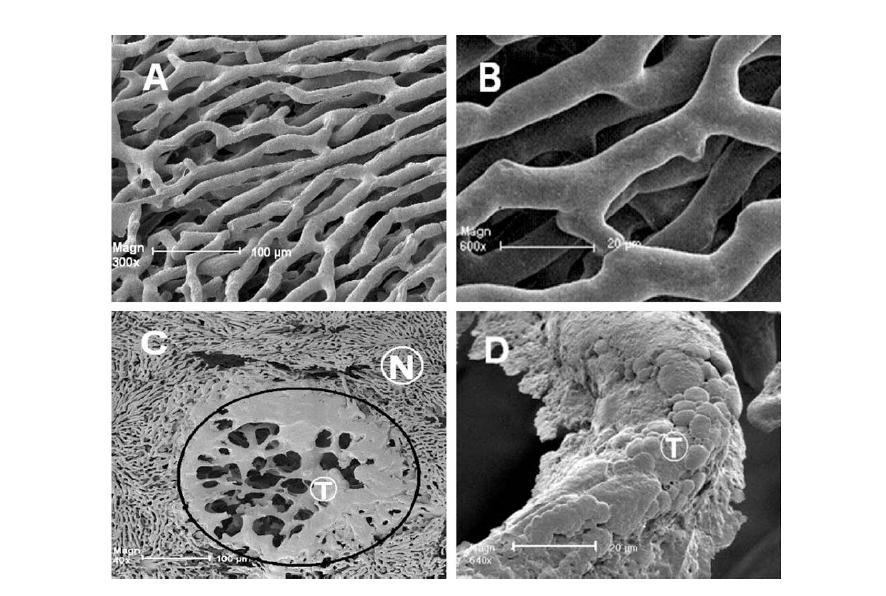
Figura 1. Micrografías de scanning electrónico de capilares sanguíneos
normales (A y B) y tumorales hepáticos (C y D). B y D son imágenes
ampliadas de la red de capilares de A y C. Mientras no hay pérdida de
polímero acrílico en el hígado normal, se observa su extravasación (por
efecto EPR) en la vasculatura tumoral (T). N es vasculatura normal.
Adaptado de [135]
2.2. iii Los nano-objetos extravasan por convección
Notablemente, en tanto las moléculas de bajo peso molecular difunden
uniformemente a través de la vasculatura hacia los tejidos, los
nano-objetos únicamente pueden extravasar a través de las fenestraciones
por convección [36]. El flujo convectivo depende del gradiente de
presión hidrostática entre el espacio intravascular (microvascular
pressure, MVP) y el espacio intersticial (interstitial fluid pressure,
IFP) y el gradiente de presión osmótica (debido a diferencias en niveles
proteicos) [37]. La funcionalidad disminuida de los linfáticos es
responsable principal de la elevada IFP, que se opone al gradiente
convectivo y a la extravasación de material hacia las células tumorales
[38, 39]. Asimismo, la elevada IFP induce un flujo intersticial radial
hacia el exterior, de entre 0,1–0,2 μm/s para un tumor aislado de 1 cm
[40] y un orden de magnitud menor para uno subcutáneo [41] (Figura 2).
Sin embargo, hay autores que consideran controvertido adjudicar la
disminución del flujo convectivo a la elevada IFP [42]. La elevada IFP
forzaría la salida de solutos y fluidos hacia la zona de menor presión
(el extremo venoso de los capilares) y no hacia el extremo arterial,
donde la presión hidrostática derivada del input cardiaco y la
resistencia vascular son mucho mayores que en el extremo venoso.
El flujo sanguíneo tumoral depende de la diferencia entre la presión
arterial y venosa, la resistencia debido a la arquitectura vascular y la
viscosidad sanguínea. En comparación a los tejidos normales, los tumores
tienen similar presión arterial pero menor presión venosa [43]. En la
microcirculación tumoral, debido a la elevada permeabilidad vascular, el
flujo sanguíneo intravascular e intersticial están acoplados. La
presencia de macromoléculas en el intersticio es en parte responsable de
la elevada viscosidad sanguínea tumoral. El flujo sanguíneo en tumores
es en promedio, menor que en tejidos normales [44].
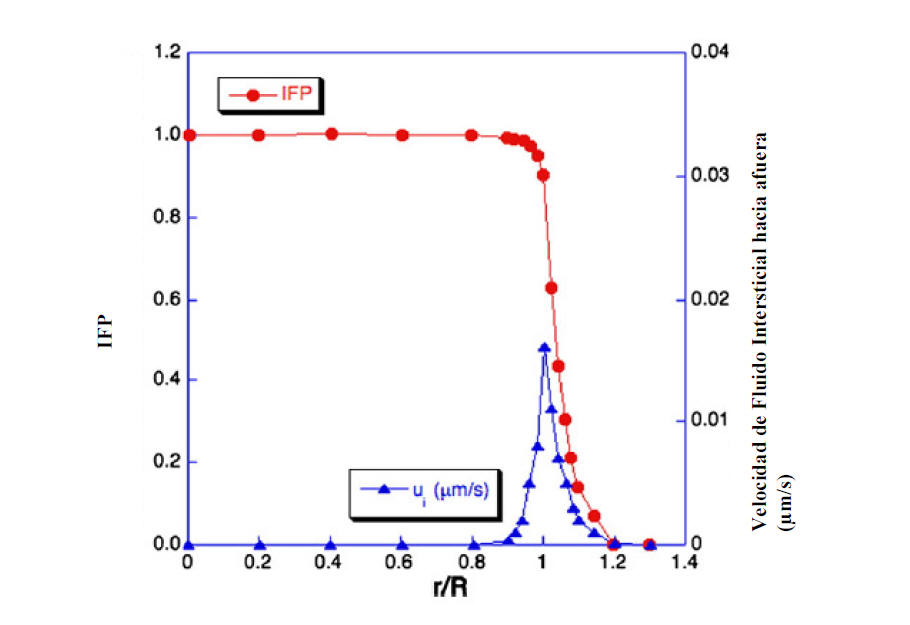
Figura 2. Presión de fluido intersticial (Interstitial fluid pressure,
IFP) y velocidad de distribución de un tumor crecido subcutáneamente. El
centro y los alrededores del tumor se indican por r/R=0 y r/R=1,
respectivamente. IFP (●) es elevada en el interior del tumor y
rápidamente decrece en la periferia. Debido al gradiente de presión, se
induce un movimiento de fluido intersticial hacia el exterior a
aproximadamente 0,02 μm/s (▲). Esta convección hacia afuera, junto con
la menor extravasación debido a la elevada IFP, conduce a un acceso
insuficiente de agentes terapéuticos [41, 61].
2.2. iv Los nano-objetos difunden con dificultad a través del
intersticio
Los tumores sólidos poseen una densa matriz extracelular (MEC),
consistente en proteínas fibrosas como colágeno y elastina y
polisacáridos como hialuronano y proteoglicanos [45], que dificultan el
ingreso de material hacia el interior tumoral. La elevada IFP tumoral
sumada al elevado contenido de glicosaminoglicanos (GAG) y colágeno
respecto de tejidos normales [46, 47], reduce el flujo convectivo
intersticial. Como resultado la difusión es el principal mecanismo de
transporte a través del intersticio. La difusión disminuye a medida que
aumenta el peso molecular y el diámetro del material [48-50]. Los
nano-objetos más pequeños son transportados a mayor distancia y con
mayor dispersión en la MEC que los de mayor tamaño, que tienden a
localizarse en el espacio peri-vascular [51].
2.3. v Heterogeneidad del efecto EPR
El efecto EPR es heterogéneo y varía sustancialmente no sólo entre
pacientes sino también entre los distintos tipos y estadios tumorales
[52, 53]. Dentro de un mismo tumor hay grandes diferencias en
permeabilidad vascular. En general, los nano-objetos (tamaño promedio de
100 nm) únicamente podrán acercarse sólo a las células de una limitada
fracción del volumen tumoral. La mayor parte del tumor apenas permite
extravasar estructuras de diámetro entre 4 y 7 nm. La distribución de la
vascularización varía con la zona y tamaño tumoral. La densidad vascular
decrece desde la periferia hacia el centro, donde se incrementan las
distancias intercapilares, que son enormes en términos de difusión de
nano-objetos. Por ejemplo, la distancia intercapilar es de unos ~50 μm
en regiones vascularizadas de adenocarcinomas mamarios de rata, pero
puede llegar a unos ~300 μm en humanos [54]. Los tumores sólidos de gran
tamaño (> 1 cm diámetro) poseen tres regiones: (a) necrótica sin
vasculatura, (b) semi-necrótica conteniendo capilares, pre-y
post-capilares y (c) establemente perfundida, con muchos vasos venosos y
pocas arteriolas (2 a 5). En general, la relación región (avascular/bien
perfundida) aumenta con el tamaño del tumor, lo que significa que el
efecto EPR es mayor en tumores más pequeños. En el interior tumoral, la
distribución de nano-objetos sería más heterogénea que la de drogas de
bajo PM, debido a que su acceso depende estrictamente de la estructura
vascular y de las serias restricciones a la difusión presentadas por el
impedimento físico de la MEC sumado a su escaso gradiente difusivo [42].
Por otro lado, la densidad vascular depende del tipo de tumor: los
carcinomas renales son muy vascularizados en tanto la densidad de
microvasculatura de carcinoma escamoso de cabeza y cuello y en carcinoma
de ovario es baja. Asimismo, la densidad vascular en algunos casos está
correlacionada con el estadio del tumor, como ocurre con el cáncer de
pulmón de células no pequeñas. Otro tipo de tumores, como el carcinoma
de células renales, no presentan correlación entre densidad vascular y
estadio tumoral. Los tumores metastásicos tienden a presentar mayor
densidad de vasculatura respecto de los no metastásicos. Diferentes
tipos tumorales secretan cantidades variables de mediadores vasculares e
inflamatorios. Por esta razón, la tasa de respuesta del sarcoma de
Kaposi (muy vascularizado y productor de grandes cantidades de VEGF) a
la monoterapia con doxorubicina liposomal peguilada (Doxil), supera
entre un 25% y un 46% a la combinación estándar del régimen ABV
(adriamicina (doxorubicina), bleomicina y vincristina) [55]. La densa
MEC propia de la mayoría de los tumores malignos pancreáticos dificulta
su irrigación; las células tumorales se alimentan por difusión de
nutrientes y oxígeno desde los tejidos adyacentes y no responden bien a
los tratamientos basados en el efecto EPR [56].
2.2.v Optimización de la estructura de nano-objetos
Para maximizar la extravasación en los sitios donde existe efecto EPR,
los nano-objetos deben circular por el máximo tiempo posible, evadiendo
la eliminación renal y la captura fagocitica. A partir de un PM de
40.000, y un diámetro hidrodinámico mínimo de 7 nm [57] (orden de peso y
tamaño de proteínas plasmáticas globulares, cuya filtración renal está
limitada por la apertura del diafragma de los pies de podocitos [58]) es
factible evitar la eliminación renal. La estabilización estérica evitará
la eliminación de los nano-objetos por fagocitosis. Empíricamente, se ha
identificado un rango de tamaño óptimopara la circulación prolongada,
extravasación y difusión de nano-objetos en tumores sólidos que
manifiestan el efecto EPR, que oscila entre los 80 y 200 nm de diámetro
[59].
El tamaño, PM y estabilización estérica sin embargo, no son los
únicos factores relevantes a la hora de lograr una óptima evasión del
reconocimiento por el sistema mononuclear fagocítico. Por ejemplo,
proteínas plasmáticas como albúmina e IgG (de PM 68.000 y 150.000,
respectivamente), se acumulan en tumores que presentan efecto EPR en
igual o mayor extensión que el primer agente antitumoral a partir del
cual se reveló el efecto EPR, el SMANCS (un conjugado de dos cadenas
copolímero estireno y ácido maleico (SMA PM 2.000) y neocarcinostatina
(NCS PM 12.000), PM total 16.000). El diseño estructural del SMANCS
dista de ser óptimo para acumularse en extensivamente en tumores
sólidos; en el tratamiento del carcinoma hepatocelular se lo inyecta
directamente en la arteria hepática y no endovenosamente. Por otro lado,
la captura hepática de ambas proteínas es sustancialmente menor que la
de SMANCS. De hecho, la mayoría de las proteínas plasmáticas
desnaturalizadas o con profundas modificaciones estructurales,
independientemente de su PM, son eliminadas muy rápidamente por
fagocitosis. Esto señala otra deficiencia en el diseño estructural de
SMANCS ya que a pesar de su bajo PM, es reconocido como ajeno en virtud
de su insuficiente biocompatibilidad.
Es importante señalar que en la inflamación asociada a infecciones,
también hay extravasación por convección de material particulado. Sin
embargo, el funcionamiento linfático normal impide su retención
prolongada. Además, el efecto EPR es “invisible” para moléculas livianas
que difunden al tumor para regresar libremente a la circulación
sanguínea. Señalemos por ejemplo que pequeñas moléculas hidrosolubles
usadas como agentes de contraste en angiografías arteriales,
experimentan un targeting pasivo en tumores, donde son retenidas unos
pocos minutos. Por el contrario, los nano-objetos que extravasan en
tumores no regresan a circulación, se acumulan gradualmente y quedan
retenidos por largo tiempo. Esta cuestión se reportó por primera vez
hace casi 30 años en roedores, donde 6 h luego de inyectar
endovenosamente Evans blue que forma un complejo con albúmina de 6 nm,
comenzó su acumulación selectiva en tumores. También se observó que la
acumulación de nano-objetos es inversamente proporcional al tamaño
tumoral. Por ejemplo, la acumulación del conjugado copolímero
hidroxipropilmetilacrilamida (HPMA)-doxorubicina, decayó de ~20% a 1–5%
dosis/g tejido tumoral, al aumentar el tamaño tanto de modelos tumorales
de ratón como xenografts humanos. En tanto la concentración plasmática
se mantuviera elevada un mínimo de 6 h, hubo un incremento progresivo de
la acumulación a lo largo de horas o días. En la clínica, luego de
administrar SMANCS en Lipiodol (SMANCS/Lipiodol) localmente vía una
infusión en la arteria hepática, se registró una veloz acumulación en el
carcinoma hepatocelular en contraste con un muy lento clearance durante
semanas. De hecho aun 2–3 meses más tarde el SMANC permaneció a razón de
20–30 µg/g tejido tumoral, una actividad remanente más de 100 veces
mayor que la concentración inhibitoria mínima del carcinoma
hepatocelular in vitro [59].
2.3. Incremento de la extravasación de nano-objetos
En promedio, tanto los conjugados polímeros-droga como drogas
liposomas como Doxil, permiten descargar un 5 % de la dosis administrada
sobre tumores que manifiestan el efecto EPR; entretanto, más del 50 % de
la dosis culmina en el hígado [60, 61]. Aunque un 5 % es un porcentaje
significativamente mayor que el acumulado como PA libre, se describen a
continuación una serie de técnicas orientadas a incrementarlo.
2.3.i Modulación del efecto EPR
El incremento de extravasación puede lograrse mediante el aumento de
la MVP, la reducción de la IFP y la normalización de la vasculatura
tumoral.
Incremento de la MVP mediante Angiotensina II: El músculo liso de la
vasculatura normal, responde a mediadores vasculares como BK,
acetilcolina, NO y calcio, ayudando a mantener constante el volumen de
flujo sanguíneo. La infusión endovenosa de angiotensina-II (AT-II),
induce hipertensión sistémica por constricción de la capa de músculo
liso. Sin embargo, ante el aumento de presión y flujo, el volumen de
flujo sanguíneo permanece constante [62-64]. En contraste, la
vasculatura tumoral no puede regular el volumen de flujo sanguíneo, que
se incrementa localmente en respuesta a la hipertensión inducida por
AT-II [59, 65]. En tumores de roedores, tras elevar la presión sistólica
mediante AT-II se observó un incremento de ~2–6 veces en el volumen de
flujo sanguíneo tumoral, dependiendo del incremento de presión [62].
Aunque su uso no se ha extendido por lo riesgoso, la hipertensión
sistémica inducida por AT II se ha aplicado en la clínica hace ya 30
años, para incrementar la acumulación de drogas de bajo PM (figura 3).
Estudios pre-clínicos en ratas mostraron que incrementando la presión
sistólica de 100 a 150 mm Hg por 15 minutos, se aumentaba 1,3-3 veces la
acumulación de SMANCS en el tejido tumoral. Además de incrementar el
efecto EPR, la vasoconstricción acompañante disminuyó el acceso de
SMANCS a riñones y medula ósea. Desde hace 10 años se reportan los
beneficios de la administración de SMANCS-Lipiodol a pacientes con
tumores sólidos (que incluyeron carcinoma hepatocelular, cáncer
metastásico de hígado, cáncer renal, colangiocarcinoma, cáncer
pancreático, entre otros) bajo un estado hipertenso inducido por AT-II.
Mediante esta estrategia es posible incrementar más de 5 veces la
concentración local de droga antitumoral, induciendo un estado
hipertenso solo por~20 min [66, 67]. Considerando que las terapias
antitumorales se llevan a cabo en o cerca de la máxima dosis tolerable,
un aumento de 2-3 veces su concentración intratumoral tendría beneficios
terapéuticos; de hecho los tiempos de regresión al 50% de la masa
tumoral se acortaron [67]. En contraste, luego de la infusión
intraarterial de PA de bajo PM no se observaron mejoras debido a su
veloz escape del tumor. No se han publicado datos acerca del empleo de
esta técnica para mejorar el delivery de Doxil u otros antitumorales
liposomales de mayor diámetro que polímeros terapéuticos.
Incremento de MVP mediante Nitroglicerina. La nitroglicerina (NG) es
un agente generador de NO, empleado por más de una centuria en el
tratamiento de angina de pecho. En el medio hipóxico (baja pO2) y de pH
ligeramente ácido del tejido cardiaco infartado, el NO2–liberado desde
NG es convertido a NO, que causa vasodilatación e incremento de flujo
sanguíneo. Dado que el medio hipóxico y acídico de los tejidos
infartados se asemejaría al de los tumores, se inspeccionó la capacidad
de NG para inducir vasodilatación e incremento local de flujo sanguíneo.
Esta estrategia incrementó 2 a 3 veces la acumulación de macromoléculas
inyectadas endovenosamente (complejo Evans blue/albúmina) en tumores,
así como la eficacia tanto de pequeñas moléculas (aclarubicina, PM 812)
como de conjugados Zn protoporfirina IX-PEG (PM 110.000). Sin embargo
estos resultados se obtuvieron luego de la administración tópica de NG
en modelos de tumores subcutáneos [68]. Esta estrategia aún no ha
llegado a la clínica. Asimismo, resta inspeccionar el efecto de la
administración de donores de NO, por rutas diferentes a la tópica.
Finalmente, aún no ha sido testeado su efecto sobre la extravasación de
nano-objetos de mayor tamaño, como el Doxil.
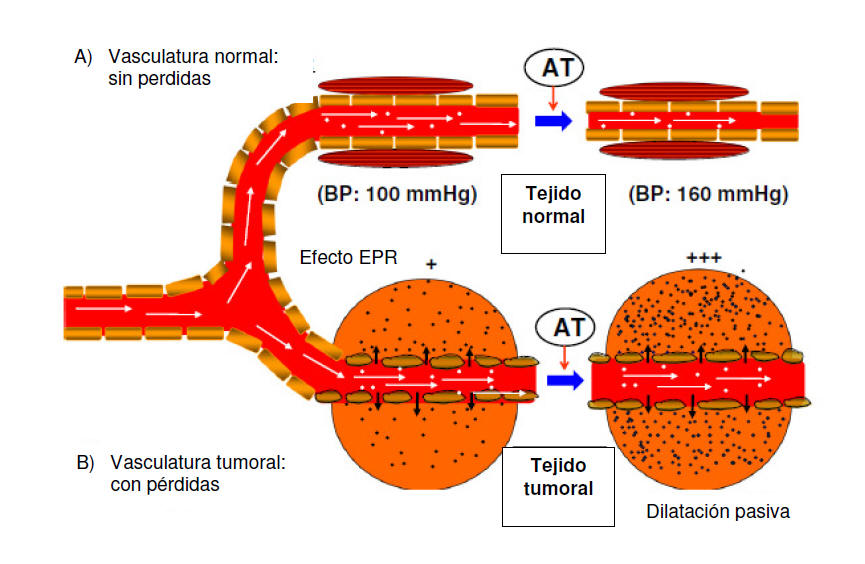
Figura 3. Representación del efecto EPR y su incremento inducido por la
hipertensión generada por angiotensina II (AT-II) y el delivery en
tejidos normales y tumorales. (A) La vasculatura normal posee una capa
de células de músculo liso que se contraen, se cierran las uniones
estrechas y se reduce el diámetro del vaso lo que resulta en menor
pérdida de contenido. (B) La vasculatura tumoral posee aberturas entre
las células endoteliales que se abren aún más al elevarse la presión
sanguínea. La hipertensión inducida por AT-II incrementa la acumulación
de drogas macromoleculares en el tejido tumoral. Adaptado de [37, 136,
137]
Incremento de MVP mediante Inhibidores de enzima convertidora de
angiotensina I (Angiotensin-converting enzyme, ACE). Los inhibidores de
ACE se emplean como agentes anti-hipertensivos y son carboxipeptidasas
que inhiben la conversión de AT I en ATII. La ATI tiene una secuencia
aminoacídica similar a la hallada en el extremo C terminal de la BK. Por
lo tanto los inhibidores de ACE potencian la acción local de la BK, el
mediador de permeabilidad vascular sobreexpresada en tumores. El
inhibidor de ACE enalapril, combinado con hipertensión sistémica
inducida por AT-II, incremento al doble el delivery de anticuerpos
monoclonales dirigidos contra cáncer gástrico sobre modelos tumorales de
ratón [65, 69]. Igual que en el caso anterior, esta estrategia no ha
llegado a la clínica y resta testear su eficacia sobre nano-objetos de
gran tamaño.
Recientemente se han testeado en roedores otras estrategias
orientadas a aumentar el efecto EPR, mediante hemo-oxigenasa 1/ monóxido
de carbono, inhibidor de TGF-β, TNF-α.
2.3.ii Incremento del delivery de nano-objetos a través del intersticio
tumoral
La difusión de nano-objetos a través del intersticio puede
incrementarse mediante a) degradación de la MEC y b) reducción de la
IFP. Aunque testeada ampliamente en pre-clínica, la utilidad clínica del
empleo de MMP (colagenasas, stromelisinas, gelatinasas y elastasas),
hialuronidasa y catepsina C (digesting decorin) para degradar la MEC e
incrementar la difusión de nano-objetos voluminosos (de alrededor de 100
nm) es discutible, ya que la digestión de la matriz no expande
sustancialmente el espacio intercelular entre células tumorales (unos 20
nm) [70, 71]. Entre otras razones, la degradación de la MEC ha sido
gradualmente abandonada porque incrementaría las chances de diseminación
tumoral [72]. También en pre-clínica, la degradación enzimática de la
MEC se ha empleado para disminuir la IFP. En este ámbito, la IFP se ha
disminuido mediante métodos físicos (hipertermia), químicos (agente
osmótico manitol) y tratamientos farmacológicos (TNF-α, paclitaxel,
radiación). Por ejemplo, la reducción de IFP luego de aplicar
hipertermia local sobre tumores subcutáneos, incrementa la extravasación
y acumulación de liposomas y vectores génicos administrados
endovenosamente (hasta 400 nm) [73, 74]. Esta estrategia sin embargo, no
ha llegado a la clínica.
Otra forma de reducir la IFP es induciendo la normalización de la
vasculatura tumoral, es decir, inhibiendo el efecto EPR. Pero aunque el
empleo de agentes anti-angiogénicos ha permitido incrementar la
extravasación de albúmina, la acumulación de estructuras de mayor tamaño
como el Doxil no aumentó y su transporte a través del intersticio de
tumores xenograft humanos se redujo [75]. En general, la inhibición del
efecto EPR mediante inhibidores de VEGF como los anticuerpos
monoclonales bevacizumab y ranibizumab puede emplearse como estrategia
antitumoral, porque bloquea el acceso de nutrientes y oxigeno; sin
embargo la misma dificulta la extravasación de nano-objetos. Hace unos
20 años comenzaron a desarrollarse inhibidores de MMP, capaces de
inhibir el efecto EPR [76]. Sin embargo, ninguno tuvo éxito clínico dada
su incapacidad para eliminar la totalidad de las células tumorales y su
toxicidad a dosis elevadas.
2.3. iii Incremento de la transcitosis: péptidos penetrantes de tumores
Recientemente se ha demostrado en pre-clínica que la
co-administración del péptido penetrante en tumores iRGD incrementó la
acumulación y el índice terapéutico de diversas estructuras,
composiciones y tamaños, tanto de pequeñas moléculas (doxorubicina),
como de anticuerpos monoclonales (trastuzumab) y nanopartículas
(Abraxane y Doxil), en múltiples modelos tumorales humanos xenograft en
ratones [77].
2.4 Utilidad de modelos pre-clínicos
Los macrófagos de ratones inmunocomprometidos producen menos VEGF,
lo que conduce a menor densidad vascular [78] y a un menor acceso de
nano-objetos al intersticio tumoral respecto de los inmunocompetentes,
que pueden acumular alrededor del doble de nano-objetos que los
inmunocomprometidos [79]. Los ratones inmunocompetentes por otro lado,
portan tumores murinos y no humanos, lo que complica la interpretación
de resultados y su extrapolación. Asimismo, la expresión de VEGF y sus
receptores en células tumorales in vitro, es mucho menos variable que en
tumores clínicos. Una diferencia sustancial entre los modelos tumorales
en animales y tumores humanos es la tasa de progresión. Los animales
usualmente desarrollan tumores clínicamente relevantes (>5 mm) una
semana después de la inoculación subcutánea de células tumorales, en
tanto un volumen tumoral equivalente puede tomar años en crecer humanos.
Un tumor de 1 g en un ratón de 30 g es aproximadamente 3% de su peso
total. En humanos, un tumor comparable pesaría 2–5 kg, una masa lejos de
ser ideal para tratarse con nanomedicinas antitumorales. Además, los
tumores implantados subcutáneamente hacen uso de la extensa red vascular
cutánea, una condición raramente hallada en tumores humanos. Los tumores
animales de rápido desarrollo producen gran cantidad de VEGF y
mediadores vasculares. Esta rápida progresión resulta en la
sobre-estimación del efecto EPR. Aparentemente, el sitio de implantación
del tumor experimental influiría en el efecto EPR. En este complejo
contexto, la utilidad como modelo predictivo de monocapas celulares y
esferoides, que carecen de vasculatura y por ende de las barreras
histológicas a la extravasación de nano-objetos, es mínima.
3. La complejidad estructural en la nano-escala
Hasta aquí hemos descripto generalidades relativas a la acción
selectiva de los nano-objetos, cuyo funcionamiento luce engañosamente
sencillo. Cabe preguntarse entonces, por que es tan lenta la aparición
de nuevos productos [80]. Las respuestas son múltiples, pero la
principal razón obedece a que la actividad de las nanomedicinas depende
estrictamente de la interacción de su arquitectura estructural en la
nano-escala con el contexto anátomo-patológico del paciente. Y sucede
que la capacidad de controlar la arquitectura de los nano-objetos a
escala industrial es limitada, además de ser muy heterogéneos los
contextos biológicos. La tecnología actual es capaz de producir en
condiciones GMP (Good Manufacturing Practice) únicamente vesículas y
nanopartículas con geometrías cuasi esféricas de lípidos (liposomas) o
polímeros naturales o sintéticos. Asimismo, las superficies pueden
recubrirse con relativa facilidad para su estabilización estérica.
Aunque no es posible obtener estructuras de mayor complejidad
estructural, este grado de simplicidad aún es muy complejo.
Analicemos la doxorrubicina liposomal peguilada (Doxil/Caelyx), uno
de los primeros nanofármacos llegados al mercado. La formulación
contiene más del 90% de los 2 mg/mL de clorhidrato de doxorubicina a pH
6.5, encapsulada en una población monomodal de liposomas de 80 nm
diámetro hidrodinámico y aproximadamente -13 mV de potencial Z. La
bicapa liposomal consiste en 3,19 mg/mL de sal sódica de
N-(carbonil-metoxi polietilenglicol
2000)-1,2-distearoil-sn-glicero-3-fosfoetanolamina (MPEG-DSPE); 9,58
mg/mL fosfatidil colina de soja hidrogenada (HSPC) y 3,19 mg/mL
colesterol. Para mantener la isotonicidad, cada mL de Doxil contiene 2
mg sulfato de amonio, buffer histidina y sacarosa. La doxorubicina se
incorpora al interior liposomal mediante el método de carga activa, para
lo que se genera un gradiente de protones al atrapar sulfato de amonio
en el interior liposomal. Este gradiente es necesario para que la base
débil doxorubicina difunda hacia el interior y quede allí retenida una
vez protonada. En este proceso, el ácido débil amonio pierde su protón y
el amoniaco resultante se elimina a través de la bicapa. La carga activa
es tan eficiente para atrapar doxorubicina, que la droga ingresada
supera su solubilidad y forma un precipitado de sulfato de doxorubicina.
Los cristales de doxorubicina responsables de la típica forma ovoide del
Doxil pueden observarse por cryo-transmission electronic microscopy
(cryo-TEM) (Figura 4). Junto con Ambisome (anfotericina liposomal),
Doxil es uno de los dos nanofarmacos más antiguos, comercializado en
Estados Unidos desde 1996. Pero aunque sus patentes estadounidenses
hayan caducado en 2010, el desarrollo de una formulación genérica de
Doxil se lleva a cabo con extremada lentitud (Lipodox, aprobado por la
Food and Drug Administration, FDA, en 2012/13 pero no por la European
Medicines Agency, EMA). La complejidad estructural de la doxorubicina
liposomal peguilada es responsable directa de su estabilidad coloidal en
circulación, farmacocinética y biodistribución. Su conocimiento
permitiría estimar la performance del producto in vivo. Sin embargo tal
complejidad es difícil no solo de estimar sino también de reproducir. A
diferencia de un fármaco convencional, conocer el origen, naturaleza,
pureza, estado cristalino y cantidad de principio activo y excipientes
no es suficiente para tener una certeza razonable acerca de la
performance in vivo de una nanomedicina [81]. Solo a titulo ilustrativo,
mencionaremos sus aspectos estructurales más relevantes: 1) los residuos
metoxi-PEG 2000 del PEG-DSPE que deben tener una conformación en cepillo
y no de hongo, para proveer máxima protección estérica; 2) constancia de
carga eléctrica superficial y potencial Z; 3) adecuado perfil de
liberación de la doxorubicina in vitro testeado bajo múltiples
condiciones, asegurando el atrapamiento la doxorubicina en el interior
liposomal frente a la dilución durante la infusión (10 ml/7000 ml
sangre); 4) gradiente de sulfato de amonio estable, ya que es
responsable de la permanencia de la doxorubicina en el interior
liposomal. Este gradiente es lábil y su disipación implica la liberación
de la doxorubicina.
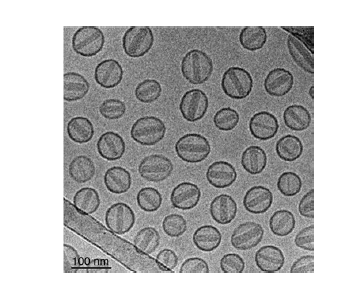
Figura 4. Cryo-TEM de Doxil.
A los efectos de aprobar una formulación genérica de doxorubicina
liposomal peguilada, la FDA [82] exige constatar la igualdad de:
condiciones internas (volumen, pH y concentración interna de sulfato de
amonio, criticas para mantener precipitada a la doxorrubicina);
morfología liposomal y cantidad de bicapas (críticas para definir carga
de droga, tasa de liberación y potencial activación de complemento);
temperatura de transición de fase de bicapas (involucrada en la
permeabilidad a la doxorubicina atrapada); tamaño de población liposomal
(critica para asegurar el targeting pasivo). Algunos de estos parámetros
son sencillos de determinar; pero otros no. Un cálculo hecho sobre la
base de la concentración de los componentes de Doxil y tamaño de
población liposomal revela que 1 ml de una dispersión comercial contiene
2,3x1014 liposomas, cada uno de los cuales acarrea unas ~10.000
moléculas de doxorubicina, de las cuales más del 95% está precipitadas
como cristales en el interior liposomal. La geometría del liposoma
influye -entre otras cosas- sobre la activación in vivo del complemento.
La técnica de cryo-TEM es en este caso la apropiada para evidenciar las
formas ovoides resultantes del precipitado interno de cristales de
doxorubicina. Las condiciones internas, como la estabilidad del
gradiente disipable o el volumen interno son aún menos sencillos de
determinar. La dificultad en predecir la performance in vivo derivadas
de la complejidad estructural en la nanoescala enlentece la aprobación
por organismos regulatorios y por ende la aplicación.
4. El targeting activo: la "bala mágica" no es real.
Hace unos 15 años, aparecieron estudios que sugerían que el targeting
pasivo podía mejorarse mediante la exhibición superficial de ligandos de
receptores selectivamente expresados por las células blanco. Sin embargo
el intento de aplicar esta estrategia conocida como targeting activo,
para incrementar el delivery de drogas mediante nano-objetos, no ha dado
los frutos esperados. Únicamente se ha implementado con relativo éxito
en fármacos consistentes en droga unida covalentemente a anticuerpos
(ejemplos: Zevalin anti-CD20 90Y-ibritumomab tiuxetan contra linfoma
No-Hodgkin), Mylotarg (anti-CD33- ozogamicin-gemtuzumab contra Leucemia,
retirado en 2010 del mercado), Ontak (anti-CD25- proteina de fusión
toxina difterica-IL2 contra linfoma de células T) y Bexxar (anti-CD20
131I-tositumomab contra linfoma No-Hodgkin).
Existen una serie de cuestionamientos sobre las estrategias de
targeting cuya selectividad depende de la interacción ligando-receptor,
surgidas tanto de la experimentación in vivo como de la clínica. Uno de
ellos se refiere a la oportunidad, cantidad y calidad de la expresión de
receptores, que pueda ser transitoria, insuficiente y/o inespecífica
[83]. En modelos tumorales de roedores, es indistinguible la acumulación
tumoral de albúmina de la de albúmina-folato. La farmacocinética y
biodistribución de nanopartículas de ácido poli- láctico-co glicólico
(PLGA) no difiere de la de nanopartículas PLGA-folato. La captura
hepática (el principal órgano de almacenamiento del exceso de folato) de
estos conjugados, explicaría estos resultados. Hasta la fecha,
tratamientos pre-clínicos y clínicos con nano-objetos derivatizados con
pequeñas moléculas como galactosamina, transferrina y folato no
produjeron resultados satisfactorios [84].
Otro de los cuestionamientos refiere a que la sobre-expresión de
receptores en tumores no sucede sincrónicamente y no conduce
necesariamente a una mayor acumulación de nano-objetos derivatizados.
Las células normales, mucho más numerosas, pueden competir exitosamente
frente a las tumorales por la captura (como ocurre con la derivatizacion
con folato). Asimismo, la endocitosis mediada por receptores que sigue a
la interacción ligando-receptor, aunque veloz, es un proceso saturable
[85]; la tasa de reciclaje de receptores en tumores es muy heterogénea,
oscilando entre 6-20 h de acuerdo al tipo tumoral [86]. En tanto los
receptores de células tumorales tienen gran heterogeneidad en densidad y
estructura [87]; la conducta de las células tumorales de un mismo
paciente puede variar de acuerdo al estadio de su progresión [88].
Estudios pre-clínicos que emplearon nano-objetos derivatizados con
fragmentos de anticuerpos, mostraron que el anticuerpo monoclonal
anti-HER2 no media un incremento significativo de acumulación de
inmunoliposomas en tumores. Los perfiles farmacocinéticos de liposomas e
inmunoliposomas son similares y la acumulación tumoral es la misma para
ambos (7-8% de la dosis inyectada/g tejido tumoral), independientemente
de la sobre-expresión tumoral de HER2 (figura 5). La presencia de
ligandos en liposomas solo incrementa sus oportunidades de
binding/internalización de aquellos que se hallan en las vecindades de
las células tumorales. Únicamente en ese caso, la internalización de
inmunoliposomas por células tumorales que sobre-expresan HER2 fue un
orden de magnitud mayor que para liposomas control. Pero como veremos en
breve, estos resultados únicamente solo pudieron conseguirse in vitro
[89]. Lo mismo sucedió con nanopartículas de poli (DL-láctico) cubiertas
con HER2 [90]. Estudios recientes muestran que inmunoliposomas
derivatizados con ligandos empleados para targeting al cerebro, no
intermedian el acceso al parénquima cerebral in vivo [91].
Por otro lado, se ha reportado que la decoración con ligandos podría
incrementar la transcitosis de nanopartículas unos 40–50 μm (3–5 capas
celulares) más allá de la vasculatura tumoral, acercándolas al core
tumoral [92, 93]. Sin embargo, la dificultad para difundir a través de
la MEC tumoral aumenta con el diámetro hidrodinámico de los
nano-objetos, como es el caso de aquellos estericamente estabilizados y
derivatizados. Finalmente, los nano-objetos derivatizados serían
eliminados de circulación mucho más rápidamente que las proteínas
nativas o polímeros biocompatibles no modificados, lo que reduce su vida
media de circulación y posibilidades de extravasación.
Un tercer cuestionamiento se refiere al hecho de que la
incorporación de anticuerpos a la superficie de nano-objetos es un reto
estructural que aún no ha podido ser superado a escala industrial. Es
extremadamente difícil obtener una población homogénea de nano-objetos
recubiertos con anticuerpos carentes de fracción Fc (para evitar su
reconocimiento por los R-Fc en células de Kupffer) unidos al extremo del
estabilizante estérico.
El cuarto y más importante cuestionamiento, es que la derivatización
con ligandos sirve para incrementar la captura de nano-objetos, por
células que expresen los correspondientes receptores, pero no incrementa
el acceso de nano-objetos a dichas células. In vivo, la cantidad de
nano-objetos capturados por un tejido blanco depende primariamente de su
posibilidad de extravasar hacia ese tejido desde circulación sistémica.
Tal posibilidad es gobernada por los parámetros del targeting pasivo
discutidos anteriormente y no por la presencia de ligandos
superficiales.
En suma entonces, la identificación de blancos moleculares en
células tumorales no garantiza el targeting del nano-objetos
derivatizados.
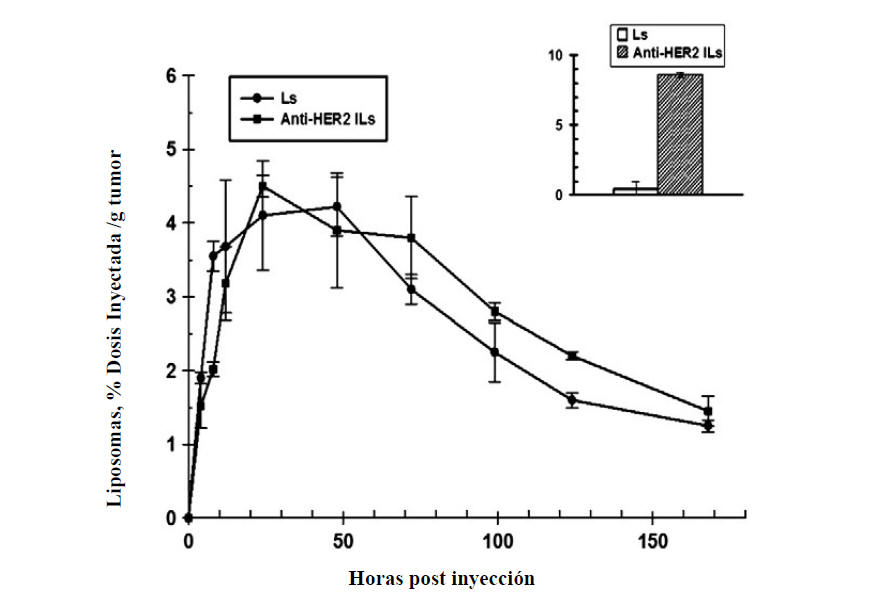
Figura 5. Farmacocinetica tumoral de inmunoliposomas anti-HER2
(Anti-HERs ILs) versus liposomas Peguilados control (Ls) en xenografts
de tumor de mama BT-474 implantado s.c. en ratones nude. Inset: captura
de immunoliposomes anti-HER2 versus liposomas control (Ls) en células de
tumor de mama que sobre-expresan HER2 (SK-Br-3) in vitro. Adaptado de
[89].
5. Comentarios acerca de la eficacia
Luego de incorporado en nano-objetos, la farmacocinética de un PA se
modifica profundamente. A excepción de las paredes vasculares, hígado,
bazo y medula ósea, los nano-objetos fluyen sin tener contacto físico
con el resto de los órganos. Asumiendo que el PA permanece retenido en
la estructura del nano-objeto, la farmacocinética del PA es
indistinguible de la de su portador. Finalmente, aun existiendo efecto
EPR, el hígado colecta la mayoría de los nano-objetos inyectados
endovenosamente, por lo general por fagocitosis a cargo de células de
Kupffer. Asi, la doxorubicina liposomal peguilada por ejemplo, puede
fluir por la vasculatura cardiaca sin que la doxorubicina tenga contacto
físico con los cardiomiocitos [95-98]. Estudios representativos que
emplearon datos normalizados a un área corporal superficial media de 1,7
m2y un peso promedio de 70 kg, mostraron que Doxil tiene mínima
interacción con tejidos sanos, reducido clearance plasmático y menor
riesgo de cardiotoxicidad [99]. Inyectada endovenosamente a dosis entre
25 y 50 mg/m2 la doxorubicina liposomal peguilada, permanece en
circulación por largo tiempo. Son típicos su mínimo volumen de
distribución (en humanos 4L, unas 65 veces menor que la droga libre),
gran área bajo la curva plasmática (unas 300 veces mayor que la droga
libre), eliminación plasmática bi-exponencial (vidas medias 2 y 45
horas, la mayor parte de la dosis siendo eliminada en la segunda fase) y
lento clearance renal (0,1 L/hora vs 45 L /hora para la droga libre). En
presencia de efecto EPR, puede acumularse entre 4 a 16 veces más que la
forma libre en tumores sólidos. En cambio la doxorubicina en formulación
convencional accede uniformemente a todos los tejidos corporales. Además
de mielosupresión severa, náuseas, vómitos y efectos mucocutáneos, puede
causar fallo cardiaco congestivo irreversible, dada su cardiotoxicidad
acumulativa y dosis-dependiente (hasta un máximo de 550 mg/m2) [94]. En
su forma nanotecnológica la doxorubicina es más segura y permite
administrar mayores dosis acumuladas con menor cardiotoxicidad [100]. Su
eficacia sin embargo, no es mayor que la de doxorubicina libre. El
acceso de doxorubicina a células tumorales, dependerá de su lenta
difusión desde los cristales precipitados en el interior liposomal a
través de bicapas poco permeables del Doxil, ya que la corona de PEG
impide su endocitosis. Doxil está diseñado para maximizar la retención
de doxorubicina en su interior, cuya liberación en circulación
conduciría a su distribución uniforme y consiguiente cardiotoxicidad. El
control del delicado balance (retención de PA en circulación/liberación
sitio específica de PA) sin embargo, es una deuda pendiente. Actualmente
se trabaja en la modificación de nano-objetos cuya estructura responda
frente a estímulos externos sitio-específicos. Ejemplo de ello son los
liposomas termo-sensibles que liberan doxorubicina luego de incrementar
su permeabilidad en respuesta a estímulos locales [101]. Pero a pesar de
su sencillez conceptual, no se han conseguido estructuras capaces de
cumplir satisfactoriamente tal misión; por lo general hay una intensa
liberación prematura de PA. Empleando métodos analíticos que permiten no
sólo cuantificar el PA en sangre sino también la matriz liposomal y
discernir entre PA libre y PA asociado a liposomas, se ha hallado que
típicamente luego de 3 h, el 50% de los liposomas permanece circulante
pero únicamente el 10% de la doxorubicina es retenida [102]. Recientes
estudios muestran que la liberación prematura de PA desde micelas
poliméricas podría retardarse mediante el entrecruzamiento de la
estructura del core [103, 104].
Los diferentes tipos, estadios y grados tumorales tienen TD muy
heterogéneos. De acuerdo al origen tisular por ejemplo, pueden variar
entre 506–5378 días (adenomas pituitarios) [105] hasta 12 días (cáncer
de estómago) [106]. Pero en tanto la toxicidad dosis-dependiente es
invariante, los tumores de crecimiento veloz, (o de bajo TD) requerirán
un aumento proporcional de la dosis de nanomedicinas biodisponibles en
el sitio blanco. Teóricamente la amplia ventana terapéutica de las
nanomedicinas basadas en el efecto EPR [107] podría ser explotada en la
dosificación personalizada para cada paciente. Pero como describimos
anteriormente, es difícil controlar la dosificación de nanomedicinas.
Por ejemplo, el Doxil se acumula en las vecindades de las células
tumorales liberando doxorubicina, que difunde al interior celular en
función de su gradiente de concentración, proporcional a su tasa de
liberación desde el interior liposomal. Sin embargo, una alta
acumulación tumoral de Doxil no significa elevada biodisponibilidad de
doxorubicina, ya que su dificultosa liberación y bajo gradiente difusivo
pueden resultar en concentración sub-terapéutica. Al margen de este
problema, en general las vesículas entre 80-200 nm son lo
suficientemente voluminosas como para asociar cantidades importantes de
PA. Pero las nanopartículas poliméricas pueden presentar el problema de
asociar insuficiente cantidad de PA. Esto ocurrió para las
nanopartículas de conjugado polimérico copolímero HPMA-paclitaxel, que
asociaron muy baja cantidad de paclitaxel (≤ 10 %) debido a su pequeño
tamaño (12-15 nm); esto sumado a la inestabilidad de la unión éster
[108, 109] resultó en concentraciones sub-terapéuticas de droga en
tejido tumoral, que se evidenció en ensayos clínicos de fase I [110]. La
rápida liberación de PA en circulación vía hidrólisis de unión éster,
asociada a un tamaño demasiado pequeño fueron los responsables de la
veloz filtración renal del conjugado copolímero de bajo PM
HPMA-camptotecina además de la toxicidad uretral registrados en ensayos
clínicos de fase I, sin que hubiera acumulación vía efecto EPR.
6. Comentarios acerca de la nanotoxicidad
La descripción detallada de la potencial toxicidad resultante de la
interacción entre seres vivos y nano-objetos (nanotoxicidad), excede los
términos de este artículo. En general podemos señalar que la interacción
con nano-objetos biodegradables y no biopersistentes representan menores
riesgos para la salud. La biopersistencia está relacionada con la
incapacidad de células fagocíticas para procesar y eliminar el material,
lo que conlleva al estrés oxidativo, inflamación crónica, y necrosis o
generación de tumores. Asimismo, las rutas de administración tópica y
mucosas son menos riesgosas que la parenteral. Los riesgos asociados a
la ruta endovenosa surgen aun para nano-objetos biodegradables, y son
varios. Por ejemplo, la doxorubicina liposomal peguilada, puede
extravasar y acumularse a través de los capilares de la piel de la
planta de los pies y de las palmas de las manos. Estas zonas muy
vascularizadas, con pequeños capilares sujetos a presión, fricción,
microtraumatismos y elevado flujo sanguíneo, favorecen la extravasación
y acumulación de nano-objetos de larga vida media de circulación. Este
fenómeno está acompañado de toxicidad conocida como eritrodestesia
palma-planta y puede ser limitante de su dosis terapéutica [111]. Como
comentáramos anteriormente, la mayor parte de los nano-objetos
administrados endovenosamente extravasa a través de fenestraciones de
sinusoides hepáticos y esplénicos. Pero mientras las funciones
esplénicas pueden ser compensadas por otros órganos del sistema
linfático, los daños hepáticos que puedan surgir tras la administración
de nanomedicinas antitumorales representan un gran reto por superar. Por
ejemplo los nanoconstructos de cis-platino (una droga con dosis limitada
por toxicidad renal) tienen menor toxicidad renal a cambio de presentar
dosis limitada por toxicidad hepática [112].
La peguilación además, puede ser responsable de la eliminación
sanguínea acelerada (accelerated blood clearance, ABC) de nano-objetos
estéricamente estabilizados. En el fenómeno ABC, el tiempo de vida media
en circulación se acorta para la segunda dosis administrada días más
tarde, a causa de la inducción de IgM anti-PEG. Este fenómeno no solo
causa captura hepática por opsonización sino eventual activación del
complemento (C) [113, 114]. De hecho, la reacción tóxica aguda más
importante luego de la administración endovenosa de nano-objetos es el
efecto CARPA (Complement Activation Related Pseudo Allergy) [115]. Esta
es una reacción de hipersensibilidad mediada por la liberación de
factores inflamatorios desde mastocitos, iniciada por la activación del
C frente a material nanoparticulado, que no requiere de la presencia de
IgE. El distrés cardiopulmonar desencadenado por el efecto CARPA puede
ser letal en individuos susceptibles. La activación del C ocurre por
adhesión directa de C1q a superficie o a complejos antígeno en
superficie-IgG/IgM, o por adhesión de lectina de unión a manosa
(MBP)/ficolinas, o por adhesión de C3, sobre superficies. A continuación
se desencadenan diferentes cascadas hidrolíticas convergentes en C3
convertasa [116]. La activación del C por vía clásica, de lectinas o
alternativa, tiene como consecuencia una mayor eliminación de
nano-objetos, ya por mayor fagocitosis luego de la opsonización o por
formación del complejo de ataque y lisis sobre estructuras sensibles
como las liposomales. El efecto CARPA se ha observado no solo luego de
la administración de Taxol (paclitaxel solubilizado en Cremophor EL) y
de Taxotere, (Docetaxel, paclitaxel solubilizado en Tween 80), y se cree
que las micelas de surfactantes no ionicos polioxietilados Cremophor EL
y Tween 80, activadoras de C in vitro, estarían relacionadas con la
manifestación del efecto CARPA in vivo. Este efecto también se ha
manifestado ante la administración de Doxil y de liposomas que no
contienen PEG, como Ambisome. Aún se desconocen las características
estructurales de los nano-objetos capaces de gatillar la activación de
C, pero han podido establecerse ciertas generalidades que permiten
predecir la manifestación del efecto CARPA. Por ejemplo, el C se activa
sobre superficies de carga positiva y aunque las cargas negativas o el
potencial Z negativo no son gatilladoras per se de activación, la misma
es sensible a la topografía superficial de cargas negativas, como ocurre
con Doxil, liposomas de dipalmitoilfosfatidilcolina (DPPC)
metoxiPEGuilados [117]. También se activa frente a la agregación de
nano-objetos. De hecho pequeños cambios en la geometría individual de
liposomas, redundan en grandes cambios de superficie total. Por ejemplo,
cada dosis terapéutica de Doxil comprende 1013–14 liposomas, lo que
representa la infusión de una cantidad de superficie en el orden de los
cm2. La detección de morfologías heterogéneas en los viales comerciales
de Doxil (elongados, irregulares, agregados), es relevante en términos
de activación de C ya que la misma es sensible a la superficie expuesta.
Recientemente se ha demostrado que mínimas variaciones en el radio de
curvatura liposomal pueden activar C por la vía clásica (vía unión de
IgM) [118]. En este contexto se ha sugerido que la doxorubicina podría
contribuir indirectamente a la activación de C al modificar la forma y
superficie de los liposomas [119].
La intensidad del efecto CARPA puede ser disminuida reduciendo la
velocidad de infusión, diluyendo la concentración liposomal y
premedicando con dexametasona. En ciertas ocasiones el fenómeno de
taquifilaxis permite la desensibilización previa mediante placebos, como
Doxibo (idéntico liposoma pero carente de doxorubicina) en el caso de
Doxil.
Los ensayos in vitro, debido a su falta de estandarización y
variabilidad, son poco efectivos para predecir activación de C in vivo
por nano-objetos. Por otro lado, la reactogenicidad frente a la
activación de C es especie dependiente, de acuerdo a:
cerdos>perros>humanos>conejos>ratas>ratones. Por lo tanto el modelo
animal porcino es el más adecuado para el estudio pre-clínico del efecto
CARPA. Claramente, los tumores xenograft humanos implantados
subcutáneamente en ratones nude inmunodeficientes, no solo son anatómica
y fisiológicamente muy distintos de aquellos que crecen en su entorno
nativo (ortotópicos), sino que este tipo de modelo excluye la percepción
del efecto CARPA del nanomaterial. A pesar de lo universal de su empleo,
los roedores están lejos de proporcionar información predictiva tanto de
eficacia terapéutica como de la potencial toxicidad de las
nanomedicinas.
7. Conclusiones
Luego de describir los fundamentos de la actividad de las
nanomedicinas antitumorales, pueden señalarse los principales aspectos
que las diferencian de las formulaciones convencionales que emplean
pequeñas drogas hidrofóbicas y de los basados en anticuerpos:
1. La actividad de las nanomedicinas depende de su posibilidad de acceso
al sitio blanco, directamente ligada a la anatomía estructural local.
Los nano-objetos no se autopropulsionan: su biodistribución depende de
fenómenos puramente hidrodinámicos [120].2. Mientras son pequeños (<2
mm) los tumores se sirven de la vasculatura de tejidos sanos adyacentes.
Desarrollar la vasculatura anómala que permita el targeting vía efecto
EPR puede tomar entre meses y años [121, 122]. Esto subraya la
desventaja de esperar a una evolución del crecimiento tumoral para
conseguir cierta selectividad y minimizar toxicidad. La dependencia del
efecto EPR significa que los tratamientos más efectivos serian aquellos
aplicados en una ventana temporal donde existiera una apropiada
arquitectura vascular, con una mínima relación masa
(avascular/vascularizada).
3. La dependencia del efecto EPR y su heterogeneidad hace que los
estudios pre-clínicos sean poco predictivos de la eficacia de las
nanomedicinas. La dificultad para conseguir modelos in vitro e in vivo,
que remeden satisfactoriamente la arquitectura vascular tumoral, es uno
de los aspectos críticos para el desarrollo de prototipos de
nanomedicinas tumorales cuya actividad depende del efecto EPR. La
relevancia clínica del targeting de nano-objetos vía efecto EPR, dada su
inusitada variabilidad, queda por demostrar. La exploración del contexto
anatomo-patológico de cada paciente, mediante el mismo tipo de
nano-objeto que permita tanto diagnóstico por imágenes como tratamiento,
podría discernir si la futura medicación será beneficiosa o no [123].
4. La farmacodinamia de las nanomedicinas es única y muy diferente a la
de los PA en formulaciones convencionales. La presencia de PA en sangre
no es necesariamente PA biodisponible. Para acceder a las células de los
tejidos irrigados por la vasculatura, las nanomedicinas deben extravasar
y atravesar la matriz extracelular; luego serán capturadas
endociticamente o liberarán el PA acarreado en la periferia de las
células blanco. Pero las nanomedicinas pueden quedar atrapadas en
fenestraciones vasculares o en la matriz extracelular. En el primer caso
se detectara PA en sangre, que no está biodisponible. En el segundo
caso, solo mediante combinación de marca radiactiva con aislamiento de
células, microscopia confocal de fluorescencia con múltiples marcas y/o
citometría de flujo es posible distinguir in situ entre PA liberado/ PA
asociado a nano-objetos/captura celular de PA libre o asociado a
nano-objetos. Por otro lado, la cuantificación del nano-objeto o de PA
por separado no significa presencia de nanomedicina, porque el PA puede
haberse disociado del nano-objeto.
5. Tanto el precio de las nanomedicinas como su complejidad estructural
son muy elevados y la evaluación del costo-beneficio de los tratamientos
con nanomedicinas oncológicas está fuera del objetivo de este artículo.
Sin embargo, en el año 2006 se comparó la efectividad y costo-beneficio
de tratamientos de segunda línea o subsecuentes con topotecan,
doxorubicina peguilada liposomal y paclitaxel (tanto monoterapia como en
combinación con compuestos de platino), sobre pacientes con cáncer de
ovarios avanzado [124]. Se observó claramente que a pesar de no ser más
efectivos, los tratamientos con doxorubicina liposomal peguilada fueron
menos tóxicos. En tal caso, el menor costo del manejo de efectos
adversos (aproximadamente £1,289 menos que topotecan y £4 menos que
paclitaxel) contrapesa el elevado costo de la nanomedicina. La próxima
generación de nanomedicinas deberá reducir la relación costo /beneficio
no sólo disminuyendo la toxicidad sino también incrementando la
eficacia. La tecnología actual sin embargo, aún debe resolver la
selectividad del delivery de citostáticos, ajustando las dosis al tiempo
de duplicación tumoral. Para ello el targeting vía efecto EPR mediante
nanomedicinas de larga vida media en circulación, ofrece una solución
poco satisfactoria. La peguilación superficial de nano-objetos, impide
su captura y subsiguiente procesamiento intracelular, y por lo tanto
reduce su oportunidad para ingresar mediante mecanismos que
incrementarían la eficacia del delivery. La resolución de esta
controversia requiere re-ingenierizar nano-objetos para a) minimizar la
complejidad estructural, evitando la decoración superficial con ligandos
complejos y b) buscar alternativas a la estabilización estérica con PEG.
La vida media de nano-objetos en circulación puede aumentarse saturando
la capacidad de captura de las células de Kupffer. Sin embargo dicha
saturación no se ha observado para nano-objetos estéricamente
estabilizados hasta dosis de 300 mg/kg. Recientes estudios sugieren que
mediante el control de la forma, podría reducirse la fagocitosis de
nano-objetos; así su vida media en circulación aumentaría sin necesidad
de complejas derivatizaciones. Por ejemplo la fagocitosis se dificulta
para aquellos nano-objetos asimétricos cuya relación de ejes mayor/menor
sea >10 y de tamaño > 1 μm [125]. Hasta el momento sin embargo, no se
han reportado estrategias antitumorales basadas en nano-objetos no
fagocitables carentes de la clásica estabilización estérica.
Por otro lado, la imodificación ( ingenierización) de los
nano-objetos permite controlar su modalidad de captura pinocítica y
tráfico intracelular (endocitosis mediada por clatrina, caveolina, por
no clatrina/ caveolina o macropinocitosis). Estas rutas permitirían el
ingreso de una elevada cantidad de PA/tiempo, así como seleccionar el
compartimiento de delivery intracelular de PA, requerida para
incrementar expresión o silenciamiento génico, modular respuestas
inmunes o evadir mecanismos de resistencia. En pre-clínica, la eficacia
de estas estrategias es muy elevada, porque la captura pinocítica
requiere de energía, pero es independiente de la difusión (y por lo
tanto del gradiente de concentración). Esto significa que la captura
pinocítica de pocos nano-objetos transportando elevadas cantidades de PA
volcaría sustanciales cantidades de PA a compartimientos selectos del
interior celular con extrema eficacia. Una de sus aplicaciones más
promisorias de la captura pinocítica es la evasión de las bombas de
resistencia a multidrogas [126,127]. Al presente, Abraxane (paclitaxel
en nanopartículas de albúmina) es el único tipo de nanomedicina
antitumoral que opera luego de ser capturada endocíticamente por células
blanco. A diferencia de Doxil, combina una reducción de toxicidad con
incremento de eficacia [128]. Actualmente el desarrollo de nanomedicinas
que incrementen el índice terapéutico en base a cambios de trafico
intracelular de la droga se perfila como extremadamente promisorio, pero
se aún se halla en fase pre-clínica fundamentalmente contra agentes
infecciosos intracelulares [129-131]. En este marco, recientemente se ha
hallado que el control de la geometría y estructura superficial de los
nano-objetos permitiría seleccionar la ruta pinocítica de captura y
procesamiento [132]. También se ha sugerido que la geometría de los
nano-objetos podría gobernar su biodistribución al experimentar
diferentes fuerzas hidrodinámicas durante la circulación [133, 134].
La sencillez extrema está llamada a operar como próxima generación
de nanomedicinas, pero para acceder a ella todavía hace falta
desentrañar la intrincada relación entre forma, tamaño, características
superficiales del nano-objeto, hidrodinámica y modalidad de
captura/trafico intracelular. La completa biocompatibilidad de los
nano-objetos serán los principales retos estructurales para el campo de
los nanobiomateriales y sobre todo para la manufactura industrial.
En este artículo hemos dado un brevísimo pantallazo desde la
investigación pre-clínica, sobre la engañosa trivialidad detrás del
diseño estratégico de nanomedicinas antitumorales basadas en el efecto
EPR, la única estrategia conocida hasta el momento, capaz de incrementar
la selectividad del delivery de citostáticos. En el mundo desarrollado
los consorcios público-privados, sustentados por una masa crítica de al
menos 20 años de investigación previa, apuntan a acelerar la
transferencia, es decir a transformar rápidamente conocimiento académico
en productos comerciales. Estas acciones, que incluyen activamente los
respectivos Ministerios Nacionales de Salud y autoridades regulatorias,
se orientan a acelerar el acceso a pruebas clínicas, con fin de asegurar
su liderazgo comercial en el campo farmacéutico. Pero en países en
desarrollo como la Argentina, nuestra familiaridad para con la
nanomedicina es engañosa. Para elaborar un plan estratégico que oriente
a varios niveles el desarrollo de nanomedicinas, es condición necesaria
pero insuficiente la formación de recursos humanos. Además debe existir
la adecuada articulación entre academia e industria, con los organismos
de control sanitario y el Ministerio Nacional de Salud. Únicamente así
podrá saberse en qué puntos críticos un país en desarrollo como el
nuestro, sacaría provecho de desarrollar este tipo de tecnología. ¿Será
la nanomedicina un instrumento sofisticado reservado a resolver
problemas de salud del Primer Mundo? Está en nosotros responder a esta
pregunta que permanece abierta. Dirección a consultar: Asociación
Argentina de Nanomedicina (www.nanomed-ar.org.ar)
Referencias
[1] Feynman RP (1960) There's plenty of room at the bottom. EngSci
(CalTech) 23: 22–36
[2] Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough
J (2013) The big picture on nanomedicine: the state of investigational
and approved nanomedicine products. Nanomedicine 9(1):1-14
[3] Bertrand N, Leroux JC(2011) The journey of a drug-carrier in the
body: An anatomo-physiological perspective. Journal of Controlled
Release 161: 152-163.
[4] Wisse E, Jacobs F, Topal B, Frederik P, De Geest B (2008) The size
of endothelial fenestrae in human liver sinusoids: implications for
hepatocyte-directed gene transfer. Gene Ther. 15 1193–1199
[5] Jacobs F, Wisse E, De Geest B (2010) The role of the sinusoidal
cells in hepatocyte-directed gene transfer. Am. J. Pathol. 176:14–21
[6] Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE (2009)
Nanoparticle interaction with plasma proteins as it relates to particle
biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug
Delivery Rev. 61: 428–437
[7] Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA
(2008) Nanoparticle size and surface properties determined the protein
corona with possible implications for biological impacts. Proc Natl Acad
Sci USA 105:14265–14270
[8] Vonarbourg A, Passirani C, Saulnier P, Simard P, Leroux JC, Benoit
J-P (2006) Evaluation of pegylated lipid nanocapsules versus complement
system activation and macrophage uptake. J. Biomed. Mater. Res. 78A:
620–628
[9] Walport MJ (2001) Advances in immunology: complement: first of two
parts. N. Engl.J. Med. 344: 1058–1066
[10] Monopoli MP, Åberg C, Salvati A, Dawson KA (2012) Biomolecular
coronas provide the biological identity of nanosized materials. Nat.
Nanotech. 7: 779–786
[11] Allen TM (1981) A study of phospholipid interactions between
high-density lipoproteins and small unilamellar vesicles. Biochim.
Biophys. Acta 640: 385–397
[12] Rensen PC, Van Dijk MC, Havenaa EC, Bijsterbosch MK, KarKrujit J,
Van Berkel TJC (1995) Selective liver targeting of antivirals by
recombinant chylomicrons: a new therapeutic approach to hepatitis B. Nat
Med 1: 221–225
[13] Kreuter J, Hekmatara T, Dreis S, Vogel T, Gelperina S, Langer K
(2007) Covalent attachment of apolipoprotein A-I and apolipoprotein
B-100 to albumin nanoparticles enables drug transport into the brain.
Journal of Controlled Release 118: 54–58.
[14] Senior J, Delgado C, Fisher D, Tilcock C, Gregoriadis G (1991)
Influence of surface hydrophilicity of liposomes on their interaction
with plasma protein and clearance from the circulation: studies with
poly(ethylene glycol)-coated vesicles. Biochim. Biophys. Acta 1062:
77–82
[15] Folkman J (1971) Tumor angiogenesis: therapeutic implications. N.
Engl. J. Med. 285: 1182–1186
[16] Berger DP, et al (1995) Vascular endothelial growth factor (VEGF)
mRNA expression in human tumor models of different histologies. Ann.
Oncol. 6: 817–825
[17] Iyer AK, Khaled G, Fang J, Maeda H (2006) Exploiting the enhanced
permeability and retention effect for tumor targeting. Drug Discovery
Today 11: 812–818.
[18] Maeda H, WuJ, SawaT, MatsumuraY, HoriK (2000) Tumor vascular
permeability and the EPR effect in macromolecular therapeutics: a
review. Journal of Controlled Release 65: 271–284
[19] Senger DR, et al (1983) Tumor cells secrete a vascular permeability
factor that promotes accumulation of ascites fluid. Science 219: 983–985
[20] Senger DR, Perruzzi CA, Feder J, Dvorak HF (1986) A highly
conserved vascular permeability factor secreted by a variety of human
and rodent tumor cell lines. Cancer Res. 46 5629–5632
[21] Nagy JA, Chang SH, Dvorak AM, Dvorak HF (2009) Why are tumour blood
vessels abnormal and why is it important to know? Br. J. Cancer 100:
865–869
[22] Herbert SP, Stainier DY (2011) Molecular control of endothelial
cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell
Biol. 12: 551–564
[23] Roberts WG, Palade GE (1997) Neovasculature induced by vascular
endothelial growth factor is fenestrated. Cancer Research 57: 765–772
[24] Graves EE, Maity A, Le QT (2010) The tumor microenvironment in
non-small-cell lung cancer. Semin. Radiat. Oncol. 20: 156–163
[25] Fang J, Nakamura H, Maeda H (2011) The EPR effect: unique features
of tumor blood vessels for drug delivery, factors involved, and
limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 63:
136–151
[26] Matsumura Y, Maeda H (1986) A new concept for macromolecular
therapeutics in cancer chemotherapy: mechanism of tumoritropic
accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 46:
6387–6392
[27] Maeda H, Yamamoto T (1996) Pathogenic mechanisms induced by
microbial proteases in microbial infections. Biol. Chem. 377: 217–226
[28] Matsumura Y, Kimura M, Yamamoto T, Maeda H (1988) Involvement of
the kiningenerating cascade and enhanced vascular permeability in tumor
tissue. Jpn. J. Cancer Res. 79: 1327–1334
[29] Wu J et al (2001) Enhanced vascular permeability in solid tumor
involving peroxynitrite and matrix metalloproteinases. Jpn. J. Cancer
Res. 92: 439–451
[30] Okamoto T et al (2001) Activation of matrix metalloproteinases by
peroxynitriteinduced protein S-glutathiolation via disulfide S-oxide
formation. J. Biol. Chem. 276: 29596–29602
[31] Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, Maeda H
(1998) Early phase tumor accumulation of macromolecules: a great
difference in clearance rate between tumor and normal tissues. Jpn. J.
Cancer Res. 89: 307–314
[32] Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK
(2004) Pathology: cancer cells compress intratumour vessels. Nature 427:
695
[33] Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher
Y, Choi NC Mathisen D, Wain J, Mark EJ, Munn LL, Jain RK (2002)
Lymphatic metastasis in the absence of functional intratumorlymphatics.
Science 296: 1883–1886
[34] Isaka N, Padera TP, Hagendoorn J, Fukumura D, Jain RK (2004)
Peritumorlymphatics induced by vascular endothelial growth factor-C
exhibit abnormal function. Cancer Res. 64: 4400–4404
[35] Fukumura D, Jain RK (2007) Tumor microenvironment abnormalities:
causes, consequences, and strategies to normalize. J. Cell. Biochem.
101: 937–949
[36] Rippe B, Haraldsson B (1987) Fluid and protein fluxes across small
and large pores in the microvasculature. Application of two-pore
equations. Acta Physiol. Scand. 131: 411–428
[37] Maeda H, Nakamura H, Fang J (2013) The EPR effect for
macromolecular drug delivery to solid tumors: Improvement of tumor
uptake, lowering of systemic toxicity, and distinct tumor imaging in
vivo. Advanced Drug Delivery Reviews 65: 71–79
[38] Boucher Y, Jain RK (1992) Microvascular pressure is the principal
driving force for interstitial hypertension in solid tumors:
implications for vascular collapse. Cancer Research 52: 5110–5114
[39] Baxter LT, Jain RK (1990) Transport of fluid and macromolecules in
tumors II. Role ofheterogenous perfusion and lymphatics. Microvasc. Res.
40 246–263
[40] Jain RK (1987) Transport of molecules in the tumorinterstitium: a
review. Cancer Res. 47: 3039–3051
[41] Baxter LT, Jain RK (1989) Transport of fluid and macromolecules in
tumors I. Role of interstitial pressure and convection. Microvasc. Res.
37: 77–104
[42] Greish K (2012) Enhanced permeability and retention effect for
selective targeting of anticancer nanomedicine: are we there yet? Drug
Discovery Today: Technologies 9: 2
[43] Peters W, Teixeira M, Intaglietta M, Gross JF (1980)
Microcirculatory studies in rat mammary carcinoma. I. Transparent
chamber method, development of microvasculature, and pressures in tumor
vessels. Journal of the National Cancer Institute 65: 631–642
[44] JainRK (1990) Physiological barriers to delivery of monoclonal
antibodies and other macromolecules in tumors. Cancer Research 50:
814s–819s
[45] Liotta LA, Rao CN (1985) Role of the extracellular matrix in
cancer. Annals of the New YorkAcademy of Sciences 460: 333–344
[46] Aukland K, Nicolaysen G (1981) Interstitial fluid volume: local
regulatory mechanisms. Physiological Reviews 61: 556–643
[47] Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK (2000) Role of
extracellular matrix assembly in interstitial transport in solid tumors.
Cancer Research 60: 2497–2503
[48] Pluen A, Boucher Y, Ramanujan S, Mckee TD, Gohongi T, Tomaso ED,
Brown EB, Izumi Y, Campbell RB, Berk DA, Jain RK (2001) Role of
tumor-host interactions in interstitial diffusion of macromolecules:
cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. U.S.A.
98:4628–4633
[49] Ramanujan S, Pluen A, Mckee TD, Brown EB, Boucher Y, Jain RK (2002)
Diffusion and convection in collagen gels: implications for transport in
the tumorinterstitium. Biophys. J. 83: 1650–1660
[50] Alexandrakis G, Brown E, Tong RT, Mckee TD, Campbell RB, Boucher Y,
Jain RK (2004) Two-photon fluorescence correlation microscopy reveals
the two-phase nature of transport in tumors. Nat. Med. 10: 203–207
[51] Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP,
Jain RK (1998) Regulation of transport pathways in tumor vessels: role
of tumor type and microenvironment. Proc. Natl. Acad. Sci. U.S.A.95:
4607–4612
[52] Jain RK, Stylianopoulos T (2010) Delivering nanomedicine to solid
tumors. Nature ReviewsClinical Oncology7: 653–664.
[53] Bae YH, Park K (2011) Targeted drug delivery to tumors: Myths,
reality and possibility. Journal of Controlled Release153: 198–205
[54] Less JR, Skalak TC, Sevick EM, Jain RK (1991) Microvascular
architecture in a mammary carcinoma: branching patterns and vessel
dimensions. Cancer Research 51: 265–273
[55] Northfelt DW, Dezube BJ, Thommes JA, Miller BJ, Fischl MA,
Friedman-Kien A, Kaplan LD, Du Mond C, Mamelok RD, Henry DH (2005)
Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and
vincristine in the treatment of AIDS-related Kaposi's sarcoma: results
of a randomized phase III clinical trial. Journal of Clinical Oncology
16: 2445–2451.
[56] Bachem MG et al. (2005) Pancreatic carcinoma cells induce fibrosis
by stimulating proliferation and matrix synthesis of stellate cells.
Gastroenterology 128: 907–921
[57] Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi
MG, Frangioni JV (2007) Renal clearance of quantum dots. Nat.
Biotechnol. 25: 1165–1170
[58] Sarin H (2010) Physiologic upper limits of pore size of different
blood capillary types and another perspective on the dual pore theory of
microvascular permeability. J. Angiogenes. Res. 2: 14
[59] Fang J, Nakamura H, Maeda H (2010) The EPR effect: Unique features
of tumor blood vessels for drug delivery, factors involved, and
limitations and augmentation of the effect.Advanced Drug Delivery
Reviews 63: 136-151
[60] Bae YH, Park K (2011) Targeted drug delivery to tumors: myths,
reality, and possibility. Journal of Control Release 153: 198–205.
[61] Kwon K, Lee SC, Han B, Park K (2012) Analysis on the current status
of targeted drug delivery to tumors. Journal of Controlled Release 164:
108-114.
[62] Suzuki M, Hori K, Abe I, Saito S, Sato H (1981) A new approach to
cancer chemotherapy: selective enhancement of tumor blood flow with
angiotensin II. J. Natl. Cancer Inst. 67: 663–669
[63] Guyton C, Hall JE (2000) The body fluids and kidneys. In Textbook
of Medical Physiology (Guyton C, Hall JE, eds), pp. 358–382, W.B.
Saunders
[64] Greish KG et al (2003) Macromolecular therapeutics: advantages and
prospects with special emphasis on solid tumour targeting. Clin.
Pharmacokinet. 42: 1089–1105
[65] Maeda H (2012) Vascular permeability in cancer and infection as
related to macromolecular drug delivery, with emphasis on the EPR effect
for tumor-selective drug targeting. Proc. Jpn Acad. B Phys. Biol. Sci.
88 53–71
[66] Arun KI, Greish K, Fang J, Maeda H (2006) Exploiting the enhanced
permeability and retention effect for tumor targeting. Drug Discovery
Today 11: 812-818
[67] Nagamitsu A, Greish K, Maeda H (2009) Elevating blood pressure as a
strategy to increase tumor targeted delivery of macromolecular drug
SMANCS: cases of advanced solid tumors.Jpn. J. Clin. Oncol. 39: 756–76
[68] Seki T, Fang J, Maeda H (2009) Enhanced delivery of macromolecular
antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 100:
2426–2430
[69] Noguchi A, Takahashi T, Yamaguchi T, Kitamura K, Noguchi A, Tsurumi
H, Takashina K, Maeda H (1992) Enhanced tumor localization of monoclonal
antibody by treatment with kininase II inhibitor and angiotensin II.
Jpn. J. Cancer Res. 83: 240–243
[70] Nagano S, Perentes JY, Jain RK, Boucher Y (2008) Cancer cell death
enhances the penetration and efficacy of oncolytic herpes simplex virus
in tumors. Cancer Res. 68: 3795–3802
[71] Kawai M, Higuchi H, Takeda M, Kobayashi Y, Ohuchi N (2009) Dynamics
of different-sized solid-state nanocrystals as tracers for a
drug-delivery system in the interstitium of a human tumorxenograft.
Breast Cancer Res. 11(4): R43
[72] Wang J, Lu Z, Gao Y, Wientjes G Au JL (2011) Improving delivery and
efficacy of nanomedicines in solid tumors: Role of tumor priming.
Nanomedicine (Lond). 6: 1605–1620
[73] Chang E, Chalikonda S, Friedl J, et al (2005) Targeting vaccinia to
solid tumors with local hyperthermia. Hum Gene Ther. 16(4): 435–444
[74] Kong G, Braun RD, Dewhirst MW (2000) Hyperthermia enables
tumor-specific nanoparticle delivery: effect of particle size. Cancer
Res. 60: 4440–4445
[75] Tailor TD, Hanna G, Yarmolenko PS, et al (2010) Effect of pazopanib
on tumor microenvironment and liposome delivery. Mol Cancer Ther.
9(6):1798–1808
[76] Wu J et al. (2001) Enhanced vascular permeability in solid tumor
involving peroxynitrite and matrix metalloproteinases. Jpn. J. Cancer
Res. 92, 439–451
[77] Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L,
Greenwald DR, Ruoslahti E, (2010) Coadministration of a
tumor-penetrating peptide enhances the efficacy of cancer drugs. Science
328: 1031–1035
[78] Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N,
Husseinzadeh N, McFarland-Mancini MM, Drew AF (2007) Macrophages mediate
inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res.
67: 5708–5716
[79] Taurin S, Nehoff H, Greish K (2012) Anticancer nanomedicine and
tumor vascular permeability; Where is the missing link? Journal of
Controlled Release164(3): 265-275
[80] Venditto VJ, Szoka FC Jr (2013) Cancer nanomedicines: So many
papers and so few drugs!Advanced Drug Delivery Reviews 65: 80–88
[81] Barenholz Y (2012) Doxil® — The first FDA-approved nano-drug:
Lessons learned Yechezkel (Chezy). Journal of Controlled Release 160:
117-134
[82] http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatory
Information /Guidances / UCM199635.pdf, accesado 01/07/2013
[83] Lehtinen J, Magarkar A, Stepniewski M, Hakola S, Bergman M, Tomasz
R, Yliperttula M, Urtti A, Bunker A (2012) Analysis of cause of failure
of new targeting peptide in PEGylated liposome: Molecular modeling as
rational design tool for nanomedicine. European Journal of
Pharmaceutical Sciences 46: 121–130.
[84] Lammers T, Kiessling F, Hennink WE, Storm G (2012) Drug targeting
to tumors: principles, pitfalls and (pre-) clinical progress. J.
Control. Release 161: 175–187
[85] Kranenborg MH, Boerman OC, Oosterwijk-Wakka JC, Weijert MCD,
Corstens FH, Oosterwijk E (1998) Two-step radio- immunotargeting of
renal-cell carcinoma xenografts in nude mice with anti-renal
cell-carcinoma X anti-DTPA bispecific monoclonal antibodies. Int. J.
Cancer 7: 74–80
[86] Paulos CM, Reddy JA, Leamon CP, Turk MJ, Low PS (2004) Ligand
binding and kinetics of folate receptor recycling in vivo: impact on
receptor-mediated drug delivery. Mol. Pharmacol. 66: 1406–1414
[87] Capone PM, Papsidero LD, Chu TM (1984) Relationship between antigen
density and immunotherapeutic response elicited bymonoclonal antibodies
against solid tumors. J. Natl. Cancer Inst. 72: 673–677
[88] Damia G, D'Incalci M (2009) Contemporary pre-clinical development
of anticancer agents- what are the optimal preclinical models? Eur. J.
Cancer 45: 2768–2781
[89] Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB,
Marks JD, Benz CC, Park JW (2006) Antibody targeting of long-circulating
lipidic nanoparticles does not increase tumor localization but does
increase internalization in animal models. Cancer Res. 66: 6732–6740
[90] Cirstoiu-Hapca A, Buchegger F, Lange N, Bossy L, Gurny R, Delie F
(2010), Benefit of anti-HER2-coated paclitaxel-loaded
immuno-nanoparticles in the treatment of disseminated ovarian cancer:
therapeutic efficacy and biodistribution in mice. J. Control. Release
144: 324–331
[91] van Rooy I, Mastrobattista E, Storm G, Hennink WE, Schiffelers RM
(2011) Comparisonof five different targeting ligands to enhance
accumulation of liposomes into the brain. J. Control. Release 150: 30–36
[92] Ruoslahti E, Bhatia SN, Sailor MJ (2010) Targeting of drugs and
nanoparticles to tumors. J. Cell Biol. 188: 759–768
[93] Lu W, Xiong C, Zhang R, Shi L, Huang M, Zhang G, Song S, Huang Q,
Liu G, Li C (2012) Receptor-mediated transcytosis: a mechanismfor active
extravascular transport of nanoparticles in solid tumors. J. Control.
Release 161: 1005-1014
[94] Kenyon J Chemotherapy and cardiac toxicity- the lesser of two
evils. Doctors Lounge Website. Available at:
http://www.doctorslounge.com/index.php/blogs/page/14030. Accessed July
13 2013.
[95] Hamilton A, Biganzoli L, Coleman R (2002) A phase I clinical and
pharmacokinetic study of polyethylene glycol liposomal doxorubicin
(Caelyx, Doxil) at a 6-week interval in patients with metastatic breast
cancer.EORTC Ann. Oncol. 13: 910–918
[96] Gabizon A, Shmeeda H, Barenholz Y (2003) Pharmacokinetics of
pegylated liposomal Doxorubicin: review of animal and human studies.
Clin. Pharmacokinet. 42: 419–436
[97] Mross K, Niemann B, Massing U (2004) Pharmacokinetics of liposomal
doxorubicin 662 (TLC-D99; Myocet) in patients with solid tumors: an
open-label, single-dose study. Cancer Chemother. Pharmacol. 54: 514–524
[98] Gabizon AA, Shmeeda H, Zalipsky S (2006) Pros and cons of the
liposome platform in cancer drug targeting. J. Liposome Res. 16: 175–183
[99] Ewer MS, Martin FJ, Henderson C (2004) Cardiac safety of liposomal
anthracyclines. Semin. Oncol. 31: 161–181
[100] Solomon R, Gabizon A (2008) Clinical pharmacology of liposomal
anthracyclines: focus on pegylated liposomal Doxorubicin. Clin. Lymphoma
Myeloma 8: 21–32
[101] Ta T,Porter TM (2013) Thermosensitive liposomes for localized
delivery and triggered release of chemotherapy. Journal of Controlled
Release 169:112–125.
[102] de Smet M, Heijman E, Langereis S, Hijnen NM, Grüll H (2011)
Magnetic resonance imaging of high intensity focused ultrasound mediated
drug delivery from temperature-sensitive liposomes: an in vivo
proof-of-concept study. J. Control. Release 150: 102–110
[103] Lee HJ, Kim SE, Kwon IK, Park C, Kim C, Yang J, Lee SC (2010)
Spatially mineralized self-assembled polymeric nanocarriers with
enhanced robustness and controlled drug-releasing property. Chem.
Commun. 46: 377–379.
[104] Koo AN, Min KH, Lee HJ, Lee S-U, Kim K, Chan Kwon I, Cho SH, Jeong
SY, Lee SC (2012) Tumor accumulation and antitumor efficacy of
docetaxel-loaded core-shell-corona micelles with shell-specific
redox-responsive cross-links. Biomaterials 33: 1489–1499
[105] Tanaka Y, Hongo K, Tada T, Sakai K, Kakizawa Y, Kobayashi S (2003)
Growth pattern and rate in residual nonfunctioning pituitary adenomas:
correlations among tumor volume doubling time, patient age, and MIB-1
index. J. Neurosurg. 98: 359–365
[106] Takahashi Y, Mai M, Kusama S (1995) Factors influencing growth
rate of recurrent stomach cancers as determined by analysis of serum
carcinoembryonic antigen. Cancer 75: 1497–1502
[107] Garnett MC, Kallinteri P (2006)Nanomedicines and nanotoxicology:
some physiological principles. Occup. Med. (Lond.) 56: 307–311
[108] Van S, Das SK, Wang X, Feng Z, Jin Y, Hou Z, Chen F, Pham A, Jiang
N, Howell SB, Yu L, (2010) Synthesis, characterization, and biological
evaluation of poly(L-gamma-glutamyl-glutamine)- paclitaxel
nanoconjugate. Int. J.Nanomedicine 5: 825–837
[109] Duncan R, Vicent MJ, Greco F, Nicholson RI (2005) Polymer–drug
conjugates: towards a novel approach for the treatment of
endrocine-related cancer. Endocr. Relat. Cancer 12: S189–S199
[110] Meerum Terwogt JM¸ ten Bokkel Huinink WW, Schellens JH, Schot M,
Mandjes IA, Zurlo MG, Rocchetti M, Rosing H, Koopman FJ, Beijnen JH
(2001) Phase I clinical and pharmacokinetic study of PNU166945, a novel
water-soluble polymer-conjugated prodrug of paclitaxel. Anticancer Drugs
12: 315–323
[111] Saini A, Berruti A, Sperone P, Bitossi R, Tampellini M, Dogliotti
L, GorzegnoG (2006) Recall inflammatory skin reaction after use of
pegylated liposomal doxorubicin in site of previous drug extravasation.
Lancet Oncol 7: 186–87.
[112] Uchino H et al (2005) Cisplatin-incorporating polymeric micelles
(NC- 6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in
rats. Br. J. Cancer 93: 678–687
[113] Ishida T, Ichihara M, Wang X, Kiwada H (2006) Spleen plays an
important role in the induction of accelerated blood clearance of
PEGylated liposomes. J. Control. Release 115: 243–250
[114] Park K (2010) To PEGylate or not to PEGylate, that is not the
question. J. Control. Release 142: 147–148
[115] Szebeni J, Muggia F, Gabizon A, Barenholz Y (2011) Activation of
complement by therapeutic liposomes and other lipid excipient-based
therapeutic products: Prediction and prevention. Advanced Drug Delivery
Reviews 63: 1020–1030.
[116] Devine DV, Wong K, Serrano K, Chonn A, Cullis PR (1994)
Liposome–complement interactions in rat serum: implications for liposome
survival studies. Biochim. Biophys. Acta 1191: 43–51
[117] Moghimi SM, Hamad I, Andresen TL, Jörgensen K, Szebeni J (2006)
Methylation of the phosphate oxygen moiety of
phospholipid-methoxy(polyethylene glycol) conjugate prevents PEGylated
liposome-mediated complement activation and anaphylatoxin production.
FASEB J. 20: 2591–2593
[118] Pedersen MB, Zhou X, Larsen EKU, Sorensen US, Kjems J, Nygaard JV,
Nyengaard JR, Meyer RL, Boesen T, Vorup-Jensen T (2010) Curvature of
synthetic and natural surfaces is an important target feature in
classical pathway complement activation. J. Immunol. 184: 1931–1945
[119] Szebeni J, Bedocs P, Rozsnyay Z, Weiszhár Z, Rosivall L, Cohen R,
Garbuzenko O, Báthori G, Tóth M, Bünger R, Barenholz Y (2012)
Liposome-induced complement activation and related cardiopulmonary
distress in pigs: factors promoting reactogenicity of Doxil and
Ambisome. Nanomedicine: Nanotechnol, Biol, Med. 8: 176–184
[120] Ruenraroengsak P, Cook JM, Florence AT (2010) Nanosystem drug
targeting: facing up to complex realities. J. Control. Release 141:
265–276
[121] Gimbrone Jr MA, Leapman SB, Cotran RS, Folkman J (1972) Tumor
dormancy in vivo by prevention of neovascularization. The Journal of
Experimental Medicine 136: 261–276
[122] Jang SH, Wientjes MG, Lu D, Au JLS (2003) Drug delivery and
transport to solidtumors. Pharmaceutical Research 20: 1337–1350
[123] MastrobattistaE (2013)Advanced drug delivery in
motion.International Journal of Pharmaceutics 454: 517– 520
[124] Main C, Bojke L, Griffin S, Norman G, Barbieri M, Mather L, Stark
D, Palmer S, RiemsmaR (2006) Topotecan, pegylated liposomal doxorubicin
hydrochloride and paclitaxel for second-line or subsequent treatment of
advanced ovarian cancer. Health Technology Assessment 10: 1-80
[125] Champion A, Mitragotri S (2006) Role of target geometry in
phagocytosis. Proc. Natl. Acad. Sci. U. S. A. 103: 4930–4934
[126] Kunjachan S, BłauAndrzej, Makel D, Theek B, Kiessling F, Etrych T,
Ulbrich K, van Bloois L, Storm G, Bartosz G, Rychlik B, Lammers
T(2012)Overcoming cellular multidrug resistance using classical
nanomedicine formulations. European Journal of Pharmaceutical Sciences
45: 421–428.
[127] Yin Q, Shen J, Zhang Z, Yu H, LiY (2013) Reversal of multidrug
resistance by stimuli-responsive drug delivery systems for therapy of
tumor.Advanced Drug Delivery Reviews, In Press.
[128] Hawkins MJ, Soon-Shiong P, Desai N (2008) Protein nanoparticles as
drug carriers in clinical medicine. Advanced Drug Delivery Reviews 60:
876–885.
[129] Etzerodt A, Maniecki MB, Graversen JH, Møller HJ, Torchilin VP,
Moestrup SK (2012)Efficient intracellular drug-targeting of macrophages
using stealth liposomes directed to the hemoglobin scavenger receptor
CD163. Journal of Controlled Release160: 72-80.
[130] Wei H, Zhuo R-X, Zhang X-Z (2013)Design and development of
polymeric micelles with cleavable links for intracellular drug delivery.
Progress in Polymer Science38: 503-535.
[131] Xu S, Olenyuk BZ, Okamoto CT, Hamm-Alvarez SF (2013) Targeting
receptor-mediated endocytotic pathways with nanoparticles: Rationale and
advances. Advanced Drug Delivery Reviews 65(1):121-38.
[132] Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME,
DeSimone JM (2008) The effect of particle design on cellular
internalization pathways. Proc. Natl. Acad. Sci. USA 105: 11613–11618
[133] Decuzzi P, Godin B, Tanaka T, Lee S, Chiappini C, Liu X, Ferrari M
(2010) Size and shape effect in the biodistribution of intravascularly
injected particles. J. Control. Release 141: 320–327
[134] Doshi N, Prabhakarpandian B, Rea-Ramsey A, Pant K, Sundaram S,
Mitragotri S (2010) Flow and adhesion of drug carriers in blood vessels
depend on their shape: a study using model synthetic microvascular
networks. J. Control. Release 146: 196–200
[135] Maeda H, Bharate GY, Daruwalla J (2009) Polymeric drugs for
efficient tumor-targeted drug delivery based on EPR-effect. European
Journal of Pharmaceutics and Biopharmaceutics 71: 409–419
[136] Maeda H (2010) Tumor-selective delivery of macromolecular drugs
via the EPR effect: background and fture prospects. Bioconjug. Chem. 21:
797-802
[137] Kono T, Maeda H, Iwai K, Tashiro S, Maki S, Morinaga T, Mochinaga
M, Hiraoka MT, Yokoyama I (1983) Effect of arterial administration of
high-molecular-weight anticancer agent SMANCS with lipid lymphographic
agent on hepatoma: a preliminary report. Eu. J. Cancer Clin. Oncol. 19:
1053-1065
|
|
Revista QuímicaViva |