Optimización de la síntesis en pasos múltiples de un acilo pirazolidinona
Pedro E. Flores Gallardo1,2*,Craig P. Jasperse 1
1Department of Chemistry, Minnesota State University Moorhead, 1104 7th Ave. South, Moorhead, MN 56563, USA. 2Life and Physical Sciences Department, Pima Community College, East Campus, 8181 E. Irvington Rd. Tucson, AZ 85709, USA.
*e-mail: pefloresgallardo@pima.edu
Recibido 24 de julio 2012- Aceptado 7 de agosto 2013.
Resumen
La síntesis modular de pasos múltiples de acilos pirazolidinonas como (9), ha sido evaluada y optimizada. El proceso comienza con ácidos no saturados (1) ó ésteres. Aminación reducativa es empleada usando aldehídos para introducir el sustituyente alquilo N-1 (3 à 6). Una acilación N-2 nueva se lleva a cabo usando ácidos carboxílicos que puede ser hecho con el reactivo de Mukaiyama (6 à 9). RMN (a veces con supresión de solvente) y GC/MS han sido usados para supervisar el progreso y la purificación de la reacción. Las condiciones de purificación de cromatografía rápida del producto (6) fueron evaluadas. Esta síntesis modular fue desarrollada para su uso en el laboratorio de química orgánica a nivel universitario.
Palabras claves: Acilo pirazolidinona, sustituyente alquilo N-1, acilación N-2, NMR, GC/MS.
Optimization of a Multistep Synthesis of an Acyl Pyrazolidinone
Abstract
The modular multistep synthesis of acyl pyrazolidinones such as (9) has been evaluated and optimized. The process begins with unsaturated acids (1) or esters. Reductive amination using aldehydes was used for introducing the N-1 alkyl substituent (3 à 6). A novel N-2-acylation using carboxylic acids can be effected by Mukaiyama’s reagent (6 à 9). NMR (often with solvent suppression) and GC/MS have been used to monitor the progress and purities of reaction. The conditions for rapid chromatographic purification of (6) were evaluated. The modular synthesis was developed for usage in undergraduate organic chemistry laboratory.
Keywords: Acyl pyrazolidinone, N-1 alkyl substituent, N-2-acylation, NMR, GC/MS.
Introducción
La habilidad de llevar a cabo síntesis de múltiple pasos que puedan transformar simples compuestos de partida a productos de gran complejidad, es una meta importante para la química orgánica moderna1. Es así que la síntesis de múltiples pasos se ha convertido en una habilidad muy crítica en este ramo2.
Este proyecto ilustra una variedad de reacciones orgánicas y procesos fundamentales durante los cursos de química orgánica universitarios, por lo cual, el tener la habilidad de realizar una síntesis de pasos múltiples es muy importante. Esta investigación está enfocada en la formación de anillos de cinco miembros llamados pirazolidinonas, con su posterior adición de sustituyentes para la formación compleja de la molécula. Las moléculas pirazolidinonas se pueden conseguir fácilmente y son muy atractivas por el variado grupo R que contienen y también porque no tienen una quiralidad definida3. Especialmente, las moléculas acilo pirazolidinonas son de gran interés como candidatos medicinales, así como en el uso de futuras reacciones sintéticas4. Estas moléculas, han demostrado actuar como un agonista selectivo de glicina con una extraordinaria actividad neuroprotectora de modelos de animales de isquemia cerebral5.
Tal como se muestra en la ilustración (figura 1), la molécula que se investigó tiene tres variables de grupos R diferentes. Se usó una variable solamente de cada grupo R; sin embargo, se pueden utilizar otros ácidos no saturados y ésteres, lo que produciría una biblioteca diversa de nuevos productos, muchos de los cuales nunca han sido sintetizados anteriormente. Este tipo de preparación modular de bibliotecas químicas es rutinaria en la industria química y en la química medicinal. Subsecuente pruebas biológicas pueden proveer ideas en las relaciones entre la estructura y la actividad de estas moléculas6.
Los estudiantes egresados interesados en química orgánica, tienen que estar preparados pera poder ser competitivos en la industria química. Es por eso, que el objetivo principal de esta mini secuencia de investigación de múltiples pasos, es involucrar al estudiante universitario quién cursa su último laboratorio de química orgánica, en un módulo de investigación de varias semanas, en este caso una por cada etapa, donde pueda practicar reacciones orgánicas aprendidas en el transcurso de sus estudios, así como practicar con instrumentos de análisis químicos. Consiguientemente, el alumno demostrará un razonamiento crítico en procesos fundamentales de la química orgánica, entendimiento e interpretación de espectros y el desarrollo de habilidades en la química sintética.
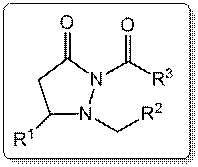
Figura 1. Ejemplo de una molécula acilo pirazolidinona con los diferentes grupos R.
Materiales y Métodos
Todos los reactivos y los solventes fueron obtenidos de la compañía Sigma-Aldrich y los materiales de vidrio de Fischer Scientific Inc. Para la purificación y caracterización se utilizó un GC/MS Agilent 5975C Series y un RMN Varian de 400 MHz.
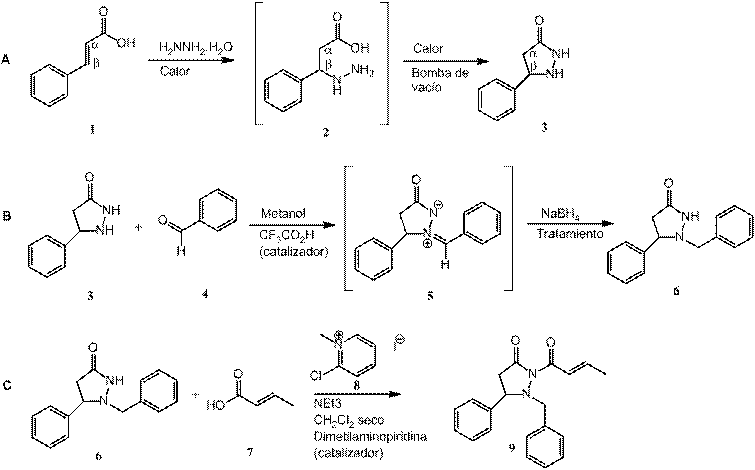
Figura 2. Esquema de la síntesis total de una molécula acilo pirazolidinona con los tres sustituyentes diferentes.
Primera Etapa (A)
En la primera etapa, 20 mmol de ácido cinámico se tratan con exceso de hidrato de hidrazina. Inicialmente, el nitrógeno se adhiere nucleofílicamente al alqueno, en un mecanismo que puede ser visto, ya sea, como un intermediario enol o un enolato. Esta reacción es hecha sin la presencia de ningún otro solvente más que el mismo hidrato de hidrazina. Siguiendo la adición, el producto 2 es sometido a la pérdida de agua y la formación de una amida cíclica con alta temperatura y bomba de vacío por 15 min, de nuevo sin la presencia de ningún solvente. La bomba de vacío es muy importante porque ayuda a destilar el agua, dado que el enlace de hidrógeno impide el cierre del anillo. Los anillos como los del producto 3 son llamados pirazolidinonas. Estas moléculas son frecuentemente muy densas y viscosas. Ambos, el intermedio 2 seguido por el calor inicial y 3 seguido por calentamiento junto a la bomba de vacío fueron evaluados. Resonancia magnética nuclear (RMN), cromatografía de gases-espectrometría de masas (GC-MS) son dos técnicas analíticas que fueron utilizadas. El compuesto en este punto es muy denso, es por eso que se usó un secador de viento caliente para introducir una muestra en el tubo de RMN y se agregó CDCl3 para disolverlo.
Segunda Etapa (B)
En la segunda etapa, al producto 3 se le añadió 10 mL de metanol para disolverlo. El nitrógeno N-1 intercambia un hidrógeno por un nuevo sustituyente de carbono, CH2R2 a través de una vía de aminación reductora usando 20 mmol de benzaldehído. En la aminación reductora de un aldehído, inicialmente un enlace C=N se forma entre el nitrógeno de la pirazolidinona y el carbono del aldehído. La carga positiva en el nitrógeno N-1 en cambio hace que el nitrógeno N-2 sea muy ácido de modo que se deeprotona en solución para dar el intermedio 5. La estructura 5 tiene dos cargas formales, pero en general es neutral. Una molécula con este tipo de situación es llamado “iluro”, comparable con el iluro de fósforo como en la reacción de Wittig que ha sido un método muy importante para la síntesis de alquenos7. El ácido es requerido para catalizar la formación del producto 5 usando 1 mmol de CF3CO2H y posteriormente fue evaluado por RMN.
El producto 5 es reducido con 24 mmol de borohidruro de sodio, el cuál provee un hidruro nucleofilico que ataca el enlace electrofílico C=N(+). El borohidruro también reacciona con el metanol para producir gas de hidrógeno y espuma. Es por eso que la reacción con NaBH4 debe ser llevada a cabo en un ambiente frío y con un contenedor con considerable espacio para la espuma generada, y con adición moderada. El borohidruro también reduce aldehídos a alcoholes; si se usa un exceso de aldehído, una cantidad significativa de alcohol puede ser producido como producto alterno contaminante el cual puede no ser removido fácilmente. Ya que el producto 6 es producido en un solvente (metanol), con mucho sodio y boro así como otros productos en los alrededores, se requiere la purificación de la solución. Muchos de los productos alternos que son creados, fueron removidos con una serie de extracciones (embudo de decantación), seguido por evaporación rotatoria, y también por embudo de vacío con alta temperatura. Dependiendo de la calidad del producto 6, se requeriría una purificación con cromatografía automatizada (“CombiFlash®”) el cual se ha convertido en un técnica fundamental en la purificación de mezclas orgánicas8.
Trabajo de aislamiento del producto (6) de la segunda etapa
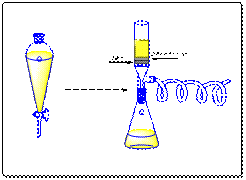
Figura 3. Proceso de aislamiento del producto (6).
De 30 mL de 2:1 éter/cloruro de metileno, solamente se añadió 20 mL en el matraz con la solución del producto 6. Posteriormente, se adicionó en el matraz 20 mL de salmuera (NaCl/agua) y 20 mL de agua destilada. Se vertió la mezcla en un embudo de decantación, y se agitó muy bien y después se escurrió la capa inferior blancuzca de salmuera. Se añadió otros 20 mL de salmuera (NaCl/agua) y 20 mL de agua destilada y se repitió el paso anterior. Se obtuvo un nuevo matraz con una junta de vidrio esmerilado y con un nuevo agitador magnético. En un embudo con frita filtrante, se añadió aproximadamente 25,4 mm de sílica e igual cantidad de sulfato de sodio. Se conectó el embudo con la frita filtrante al matraz nuevo y se conectó con la manguera de la bomba de vacío. Se escurrió la solución orgánica del embudo de decantación en el embudo con frita de filtración, y se abrió lentamente la bomba de vacío. Posteriormente, se añadieron los 10 mL restantes de 2:1 éter/cloruro de metileno en el matraz inicial y se agitó y vertió en el embudo de decantación. De nuevo, fue agitado y vertido en el embudo con frita filtrada. El matraz con la solución se evapora rotatoriamente por 5 min o hasta que la solución concentrada. Se añadieron 10 mL de cloruro de metileno en el matraz con solución concentrada y se agita y calienta a 190 oC en la placa de calentamiento conectada a la bomba de vacío por 10 min. Se desconectó la bomba de vacío y añadieron otros 10 mL de cloruro de metileno para evaporar el metanol completamente y después se conectó de nuevo la bomba de vacío, agitando y calentando por otros 5 min hasta que el solvente se evaporara completamente. Una muestra fue analizada por RMN y GC/MS.
Tercera etapa (C)
Basados en los milimoles obtenidos del producto 6, se le adicionaron 1,2 equivalentes de ácido crotónico, 1 mmol de 4-dimetilaminopiridina, 1,4 equivalentes del reactivo de Mukaiyama y finalmente 2,8 equivalentes de trietilamina como catalizador. El producto acilo pyrazolidinona 9 es formado en solvente y acompañado por múltiples productos alternos. La purificación envolvió extracción líquido-líquido (embudo de decantación) y evaporación rotatoria, siguiendo los mismos pasos que el proceso de aislamiento del producto 6, solamente cambiando NaCl/agua por HCl/agua. Cromatografía CombiFlash® automatizada podría ser necesaria, dependiendo en que tan puro o impuro sea el producto. Posteriormente, RMN y GC-MS fueron usados de nuevo para supervisar las muestras crudas y purificadas.
Resultados y Discusión
Caracterización y Purificación
El producto 3 es evaluado por cromatografía de gases y espectrometría de masas. Este paso es hecho para que el estudiante pueda tener una certeza de que su síntesis en su primera etapa, haya sido completada satisfactoriamente. Usualmente se le recomienda al estudiante que haga cálculos de masa molecular del producto a ser evaluado, para que posteriormente pueda ser comparado con los datos obtenidos con el espectrómetro de masas. Eventualmente, también son evaluados los productos de interés a como se vaya completando la síntesis total.
El mismo caso aplica para el uso de resonancia magnética nuclear. El uso de esta técnica tiene una ventaja sobre GC/MS o cualquier otra técnica de espectroscopía, ya que es un método analítico que revela la identidad y la pureza de cualquier compuesto orgánico9. Los H1 RMN de los productos 3, 6 y 9 son identificados como materiales limpios y libres de solvente. Esos espectros son los más fáciles de analizar en detalle. Para los espectros H1 RMN de los intermediarios 2 y 5, puede ser observado considerablemente la presencia de solvente, lo cual puede interferir o superponerse con partes o muchas de un espectro. Afortunadamente, la mayoría de los solventes usados en la espectroscopia RMN orgánica están disponibles en forma deuterada. Sin embargo, muchas moléculas de interés bioquímicos y medicinal usan solventes que no se encuentran en dicha forma10. El H1 RMN del experimento del intermediario 5, se usa una metodología de supresión de solvente de metanol para mejorar la visualización de los hidrógenos reales en la muestra.
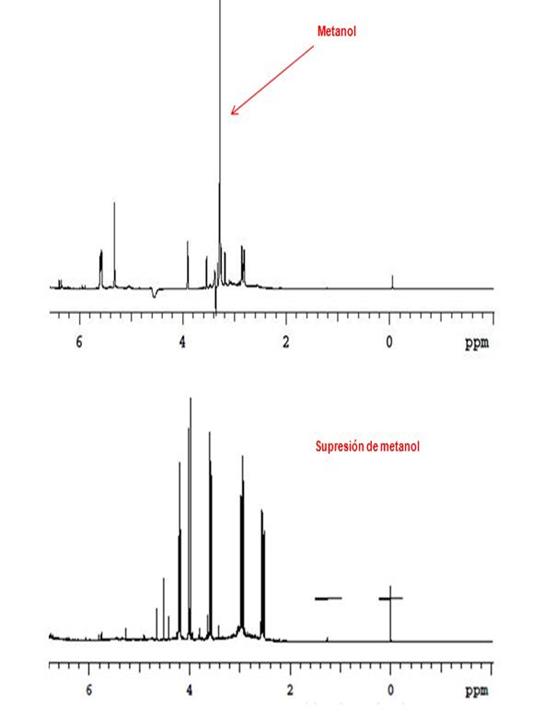
Figura 4. Espectro de H1 RMN del producto 5 con y sin supresión de solvente (me
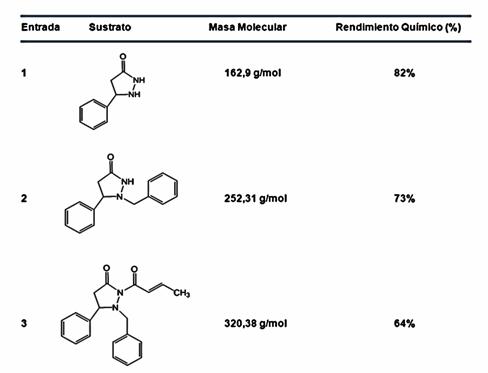
Figura 5. Rendimiento químico porcentual de los mayores productos obtenidos.
Conclusión
El porcentaje de rendimiento químico total obtenido de la síntesis fue de un 38%, lo que dio lugar a que los pasos optimizados dieran un rendimiento químico de mejora de un 8% en promedio. El uso de metanol dio un incremento en el rendimiento total en comparación al uso de etanol que era previamente registrado. El metanol es mucho más fácil de evaporar y concentrar que el etanol, además, es mucho más fácil de suprimir en RMN para un análisis más claro. La prueba de las bombas de vacío, demostraron que el rendimiento de la síntesis es dependiente de tener un sistema cerrado (no expuesto al aire libre) para el lapso indicado o de otra manera el trabajo de otros estudiantes se vería afectado considerablemente al llevar a cabo las reacciones. Los constantes análisis químicos de RMN y GC/MS de los todos los compuestos derivaron en tener un mejor entendimiento de las reacciones y la purificación de los compuestos, algo que no se había registrado minuciosamente con anterioridad.
Las reacciones y técnicas de química orgánica de esta investigación no están determinantemente dirigidas para estudiantes universitarios, sino para investigadores también, ya que se ha tocado tópicos de gran interés de la química farmacéutica donde se crean bibliotecas de moléculas complejas que pudiesen tener actividad biológica significativa.
Referencias
(1) Duff, D. B.; Abbe, T.G. and Goess, Brian C. A Multistep Synthesis Featuring Classic Carbonyl Chemistry for the Advanced Organic Chemistry Laboratory. Journal of Chemical Education. 89, 406-408, 2012.
(2) Utku, Y.; Rohatgi, A.; Yoo, B.; Kirshenbaum, K. Rapid Multistep Synthesis of a Bioactive Peptidomimetic Oligomer for the Undergraduate Laboratory. Journal of Chemical Education. 87(6), 663-639, 2010.
(3) Sibi, M.; Stanley, L.; Xiaoping, N.; Venkatraman, L.; Liu, M; Jasperse, C. The Role of Achiral Pyrazolidinone Templates in Enantioselective Diels-Alder Reactions: Scope, Limitations and Conformational Insights. Journal of the American Chemical Society. 129(2), 395-405, 2006.
(4) http://www.web.mnstate.edu/jasperse/Chem365/Chem365.html, accesado 15/05/2012.
(5) Di Fabio, R; Araldi, G.; Baraldi, D.; Cugola, A.; Donati, D.; Gastaldi, P.; Goacobbe, S. A.; Micheli, F.; Pentassuglia, G. Synthesis and Pharmacological Characterization of a Conformationally Series of Indole-2-Carboxylates as in Vivo Potent Glycine Antagonists. Farmaco. 56(10), 791-798. 2001.
(6) Jasperse, C.P. Multistep Synthesis of an Acyl Pyrazolidinone, Multistep Synthesis Research Module version 2010. 51, 2010.
(7) Wang, Z.; Zhang, G.; Guzei, I.; Verkade, J.C. PhCH=P(MeNCH2CH2)3N: A Novel Ylide for Quantitative E Selectivity in the Wittig Reaction. Journal of Organic Chemistry. 66, 3521-3524, 2001.
(8) Butler, J.; Choung, W.; Kurth, M. Flash Chromatography: A Novel Pressurization Apparatus. Journal of Chemical Education. 87(11), 1265, 2001.
(9) Yang, B. Analysis and Purification Methods in Combinatorial Chemistry. John Wiley & Sons, Inc. p. 3. 2004.
(10) Claridge, T.G.W. High-Resolution NMR Techniques in Organic Chemistry. Tetrahedron Organic Chemistry. 27, p. 354, 2009.
|
|
Revista QuímicaViva |