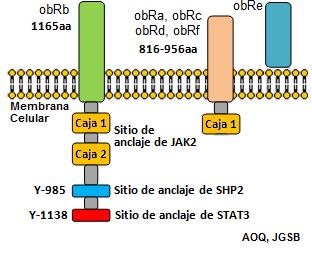
En los humanos, los receptores de leptina (isoforma larga obRb) presentan tres
residuos de tirosina (985, 1077 y 1138) en el dominio intracelular, cuyas
secuencias de aminoácidos circundantes a estos, proveen de un sitio de anclaje
para una proteína de señalización en particular. El residuo de tirosina 1138
ancla a la proteína traductora de señal y activadora de transcripción STAT3,
mientras que el 985 ancla a SHP2 como se muestra en la figura 2. (20) lo que
lleva a la activación de la vía ERK(21-23). Fosfo-Tyr985 también se une a SOCS3,
que regula negativamente la señalización de la leptina (24). Fosfo-Tyr1077 se
une a la señal del transductor y activador de transcripción STAT5 (25, 26).
Fosfo-Tyr1138 recluta STAT3, permitiendo así que JAK2 fosforile y active
STAT3(22).También esta isoforma presenta en la región intracelular dos regiones
conocidas como caja 1 y 2, las cuales anclan proteínas JAK2 las cuales son
fundamentales para la activación de STAT3.
Tabla 1. Isoformas generales de receptores de leptina
|
Isoforma |
Función |
Localización |
|
Larga
(obRb) |
Transducción de señales citoplasmáticas |
Hipotálamo |
|
Corta
(obRa) |
Transporte intracelular y degradación de leptina |
Hipotálamo, placenta, ovario. |
|
Soluble
(obRe) |
Regulación de los niveles de leptina en plasma. |
Circulación sanguínea |
Las isoformas que contienen dominios intracelulares cortos reclutan efectores
que regulan negativamente la vía de señalización JAK2/STAT3 mediada por leptina
y son incapaces de activar una vía de señalización a pesar de ser expresados en
muchos tejidos, por lo que su función aún no está esclarecido aunque existe
evidencia de que estas isoformas pueden estar involucradas en el transporte
intra- y extracelular de la leptina.(9)
Finalmente la isoforma soluble obRe, que a pesar de no presentar la capacidad de
activar vías de señalización, se ha visto que su función principal es la de
controlar los niveles de leptina en suero, debido a su gran afinidad por la
leptina que la secuestra previniendo la activación de los receptores de leptina
regulando a si sus efectos en las células blanco.(5)
La leptina induce señales que comprenden muchas vías de señalización, las cuales
son dirigidas por varias citoquinas (por ejemplo, la vía canónica, en la que
está involucrado: JAK2/STAT, MAPK/ERK1 /2 y PI-3K/AKT1 y las vías de
señalización no canónica: PKC, JNK y cinasa MAP p38). Cada una de estas señales
que induce la leptina es esencial para los efectos biológicos de la ingesta de
alimentos, balance de energía, adiposidad, sistema inmune y endocrino así como
en la carcinogénesis.(27)
La vía de JAK/STAT es principalmente activada por interferones, interleuquinas y
otras citoquinas cuyos receptores pierden actividad intrínseca como cinasas y
comprenden una familia de cuatro receptores de JAK y siete factores de
transcripción (STATs) de 85 a 95 kDa que son regulados por fosforilación en
residuos específicos de serina y tirosina. (28) Sólo JAK2 es activado durante la
señalización del receptor de leptina ob-R. Al final de la señalización de ob-R
por la leptina, JAK2 se une a STAT3 y activa STAT1, STAT5 y STAT6, por lo tanto,
el reclutamiento de STAT proviene de la fosforilación de tirosina por JAKs, con
la consecuente disociación de del receptor y la formación de homodímeros o
heterodímeros, que en cáncer mamario se explicará más adelante. (29)
La vía del sistema celular MAPK que puede ser estimulada por la forma corta o
larga del ob-R, es posible que sea activada por dos diferentes maneras, vía
fosforilación de tirosina del receptor de JAK2 o independientemente de la
fosforilación del receptor. En monocitos la leptina induce expresión y secreción
del receptor antagonista de IL-1usando la vía MAPK que activa NF-B.(30)
Se observa también que la leptina comparte con otras citoquinas, factores y
estresores de crecimiento, la habilidad de activar o estresar la proteína cinasa
c-Jun N terminal (JNK). Por ejemplo la leptina aumenta la producción del TNF-
vía p38 y JNK MAPK (30). Entre los posibles blancos terapéuticos para inhibir la
activación de p38 y JNK MAPK por inducción de la leptina, parece que la
regulación del factor de transcripción NF-B sería un importante regulador de
citoquinas proinflamatorias tales como TNF- e IL-1 (28)
La activación del receptor de la leptina también activa la vía de
fosfatidilinositol 3-quinasa (PI3K). Si bien algunas de las vías de señalización
inducidas por leptina, tales como la vía JAK2/STAT3, inducen respuestas
celulares principalmente a través de cambios en la expresión de genes, la vía
PI3K afecta a las propiedades celulares más rápidamente, a través de cambios
post-traduccionales tales como la fosforilación de proteínas.(31)
Las fosfatidilinositol 3-kinasas (PI3Ks) son complejos heterodiméricos
compuestos de subunidades reguladoras y catalíticas que reclutan lípidos como
segundos y controlan una amplia variedad de funciones celulares, tales como
supervivencia, crecimiento, metabolismo y quimiotaxis.
El complejo PI3K fosforila el grupo 3-hidroxilo del anillo inositol en sustratos
lípidos conocidos como fosfatidilinositoles (PtdIns). La actividad de PI3Ks es
contrarrestada por enzimas fosfatasas que específicamente remueven residuos
fosfato de los PtdIns. Un ejemplo es la phosphatase and tensin homolog, deleted
on chromosome ten (PTEN), que desfosforila el grupo 3-hidroxilo del anillo de
inositol.(31)
La vía de señalización PI3K es inducida por varios factores de crecimiento y
otras hormonas. Por ejemplo, la activación del receptor de insulina provoca la
fosforilación de los residuos de tirosina en el sustrato del receptor de
insulina-1 y 2 (IRS-1 y el IRS-2). Esta fosforilación induce una rápida
asociación entre el IRS-1 o IRS-2 con PI3Ks, causando la subsiguiente activación
del complejo PI3K (32). La fosforilación de PtdIns inducida por PI3K activa
blancos intermedios, tales como Akt (también conocida como proteína kinasa B o
PKB) y fosfoinosítidos dependiente de la quinasa 1 (PDK-1), que, a su vez,
transmiten y coordinan la mayoría de los efectos intracelulares inducidos por la
insulina.
La señalización PI3K es una importante vía que relaciona la obesidad, la leptina
y el aumento del riesgo de cáncer de (33). Sin embargo, otras vías
intracelulares, incluyendo JAK/STAT3 y MAP, también pueden estar implicadas en
la inducción de cáncer por leptina.(34)
La glucólisis provee una condición permisiva para la leptina para estimular la
vía de JAK2/STAT3. AMPK parece ser un regulador negativo de la señalización de
la leptina. La glucosa estimula la señalización de leptina al menos en parte al
inhibir la capacidad de AMPK para suprimir la señalización de la leptina (Su H,
Jiang L, 2012).
La leptina y la IL-1 están asociadas en diversas situaciones patológicas(35,
36), sugiriendo una interacción entre ambas. Más aún, la leptina regula a los
miembros de la familia IL-1 en un contexto de diabetes (37) y en células de
cáncer de endometrio (37, 38). La leptina incrementa los niveles de proteína y
ARNm de todos los componentes del sistema IL-1 en líneas celulares de cáncer
mamario de ratón. Además, la regulación por leptina del promotor IL-1α involucra
la activación de los factores de transcripción SP1 y NF-κB.(39)
Expresión de receptores de leptina en células neoplásicas
Receptores de leptina han sido identificados en células malignas de pulmón,
estómago, glándula mamaria, colon y en células leucémicas. En estudios in vitro
se ha encontrado este receptor en líneas celulares de cáncer de pulmón escamoso
(SQ-5) y de cáncer de colon (HT29). Específicamente, son diversos los estudios
que por inmunohistoquímica han revelado la sobre-expresión de receptores de
leptina obR en muestras de tejido de cáncer de mama de diferente estadio, desde
primario hasta metastásico. (1, 5, 12, 13, 40) Con lo antes expuesto podemos
concluir que la leptina ejerce su efecto biológico en las células neoplásicas a
través de los receptores obR presentes en dichas células.
Vía de señalización en cáncer mamario
La vía de señalización JAK/STAT en general participa en la regulación de la
proliferación, sobrevivencia, motilidad y apoptosis celular en diferentes
órganos.(41) Existen diferentes tipos de proteínas “JAK´s” (JAK1-3 y Tyk2) así
como diversas proteínas “STAT´s” (STAT 1-4, STAT 5 a-b y STAT 6). JAK1 y 2 junto
con STAT3 y 5 son las que más se han encontrado sobre-expresadas en diversas
células cancerosas (en específico cáncer mamario) tanto in vivo e in vitro, de
hecho hoy en día se considera al gen que codifica a STAT3 como un oncogén debido
a que su sobre-expresión regula la transformación oncogénica de diversas
células.(42)
La leptina ejerce sus efectos biológicos a nivel celular a través de la vía de
señalización JAK2/STAT3, sin embargo el receptor obR vía JAK2 y Grb2 puede
activar otras vías como la ERK, Akt, IRS-1, la AP-1 (Transcription Activator
Protein 1) y la SOCS3 que regula los efectos de la leptina en las células.(9,
12) Un estudio realizado por Dan Wang, de la Universidad de Pekín China,
mediante el empleo de los inhibidores AG490 y U0126 de las vías JAK2/STAT3 y ERK
respectivamente, demostró que el bloqueo de la vía JAK2/STAT3 produce una mayor
inhibición en la proliferación de la línea celular MCF-7.(1) Diversos estudios
realizados han permitido establecer que la leptina promueve el desarrollo y
progresión de las células neoplásicas activando la vía JAK2/STAT3 en estas
células.
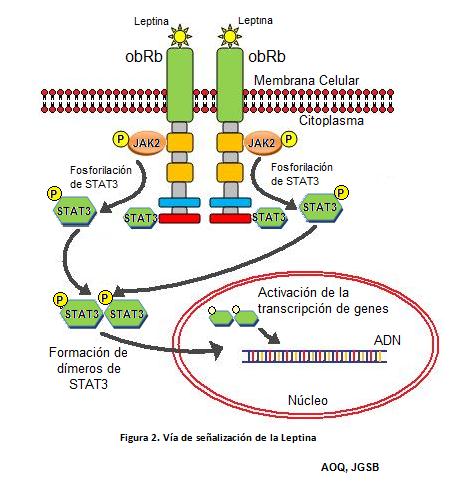
El mecanismo de señalización comienza cuando la leptina se ancla a su receptor
en una relación 1:1, dicho anclaje cambia la conformación tridimensional del
receptor el cual afecta la subestructura FERM en la proteína JAK2 anclada al
receptor del lado intracelular, dicho cambio en FERM permite que JAK2 se active.
Cabe mencionar que el receptor de leptina no posee actividad enzimática por lo
que necesita de otras proteínas para activar diversas vías de señalización
citoplasmática. La JAK2 activada rápidamente fosforila el residuo de tirosina
1138 en el dominio citoplasmático del receptor. Esta fosforilación provee de un
sitio de anclaje para la proteína STAT3 que es reclutada a través de su dominio
SH2. Ya unidas las proteínas STAT3 con el receptor, son fosforiladas por JAK2.
Como se encuentra formado un complejo con varios receptores de leptina, esto
permite el anclaje de más proteínas STAT3 que son activadas por más JAK2. Estas
fosforilaciones inducen que las STAT3 formen dímeros a través de sus residuos de
tirosina fosforilados y sus dominios SH2, estos dímeros se liberan del sitio de
anclaje en el receptor y con ello permiten su translocación al núcleo donde
modulan la transcripción de diversos genes blanco. (9, 12, 40, 43)
Mecanismo molecular
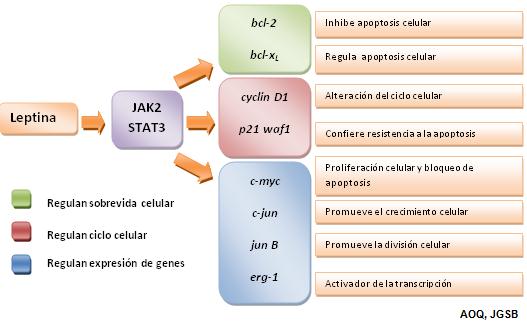
En el núcleo los dímeros de STAT3 actúan como activadores de transcripción de
diversos genes blanco, como: c-myc, cyclin D1, p21 waf1, c-jun, junB, erg-1 y
Bcl-2, los cuales están fuertemente involucrados en el crecimiento y
proliferación celular. La figura 3 resume la función de cada uno de estos genes
en el desarrollo y progresión de las células neoplásicas. (44-52)
Así también los dímeros de STAT3 pueden activar genes de SOCS3 que están
involucrados en la modulación de los efectos que tiene la leptina en las células
y genes del (VEGF) que están involucrados en la angiogénesis. (9, 41-43) También
se sabe que STAT3 interactúa y recluta a miembros de la familia de coactivadores
p160 como SRC-1, pero no GRIP1 y ABI1 a sus sitios promotores de transcripción
de genes. (1) La leptina también interactúa con miembros de la familia de IL-1,
ambas conocidos como adipocitoquinas ya que son secretadas por el tejido
adiposo, pero también por tejido epitelial del tumor mamario. De acuerdo con
esto último, Perrier generó en su estudio una hipótesis, que las células
adiposas mamarias expresan como agonistas a la IL-1 y la leptina y como
antagonistas al receptor a de IL (ILra) y la adiponectina.
 (53)
(53)
Figura
3. Genes activados por leptina vía JAK2/STAT3. c-myc codifica para una proteína
MYC que está involucrada en la regulación de la apoptosis mediante la alteración
del ciclo celular en la fase S. cyclin D1 forma complejos con CDK4 y CDK6 lo que
aumenta al factor E2F en la células permitiendo el paso de fase G1 a S. p21 waf1
induce resistencia a la apoptosis. c-jun activa genes que promueven el
crecimiento (BEX2). jun B afecta los niveles de ciclina A2 en la fase G2 que
finaliza en defectos en la regulación de la mitosis celular. erg-1 activa
diferentes factores de crecimiento y angiogénesis (VEGF). Bcl-2 inhibe la
apoptosis celular evitando que el ADN dañado funcione como una señal de
activación de apoptosis y bloqueando genes implicados en esta. (44-52)
La regulación negativa de la vía de señalización de leptina se da a través de
las proteínas supresoras de señalización de citoquinas (SOCS). Entre estas, la
SOCS3 a través de su dominio SH2, se une a la JAK2 activada o con los residuos
de tirosina fosforilados en el receptor obRb evitando la activación de JAK2 con
lo cual es capaz de inhibir la vía de señalización por leptina. (9)
Leptina en carcinogénesis mamaria
Los niveles elevados de leptina presentes en pacientes con obesidad, representa
la causa por la que esta enfermedad sea un factor de riesgo muy importante en el
desarrollo de cáncer de mama, así como su progresión. Hablando sobre los
receptores de leptina en las células neoplásicas del tejido mamario, se ha
demostrado que en dichas células están sobre-expresados estos receptores,
principalmente obRa y obRb en comparación a las células normales. Cabe mencionar
que las otras isoformas cumplen diferentes funciones cuyos efectos convergen en
la estimulación del desarrollo del cáncer de mama, pero dichas funciones aún
continúan bajo estudio. La presencia del receptor obRb permite a la leptina
activar la vía de señalización JAK2/STAT3, que induce fenómenos de inmortalidad
(inhibición de la apoptosis), proliferación e invasión celular reflejándose en
la inducción de la transformación y progresión de las células neoplásicas.(1,
12)
La sobreexpresión tanto de leptina como de sus receptores ha sido demostrada en
diversos estudios utilizando inmunohistoquímica y técnicas de biología molecular
como la reacción en cadena de la polimerasa en tiempo real (RT-PCR) en células
de cáncer primario y metastásico de mama. En el 2009, en Francia, Jardé y
colaboradores, encontraron una sobre-expresión de leptina en la línea celular
MCF-7, además, demostraron que la exposición de estas células a leptina induce
la expresión de ARNm tanto de leptina como de sus receptores.(54) Estos
resultados indican la capacidad de la leptina de auto-regular la amplificación
de su actividad en las células mediante el aumento de la expresión de los
componentes necesarios para su señalización celular.
En relación a la inducción de metástasis en células neoplásicas, Ishakawa en
2004 en un estudio realizado en Japón, reveló que de los cánceres de mama
estudiados que fueron positivos para receptores de leptina, el 34% presentó
metástasis avanzada, mientras que aquellos que carecían de dichos receptores no
presentaron metástasis (55). Estos resultados permiten inferir la función de la
leptina y sus receptores como moduladores de la proliferación celular hacia
otros sitios del cuerpo. En adición, un estudio realizado por Revillon en el
2006 demostró que niveles elevados de ARNm de isoformas cortas de receptores de
leptina estaban asociados a una disminución del tiempo en el cual pacientes
diagnosticados con cáncer de mama, presentaban una remisión después de la
cirugía de extirpación de la masa tumoral. (56)
En relación al género, los niveles de leptina son mayores en mujeres que en
hombres. Debido a la diferente regulación de leptina por las hormonas sexuales,
como los estrógenos que inducen incremento en la secreción de leptina y la
testosterona que disminuye los niveles de esta, se infiere un aumento a la
susceptibilidad de desarrollar cáncer mamario en el sexo femenino. Además, se ha
visto que la leptina aumenta la actividad productora de estrógenos en el tejido
adiposos en mujeres post-menopáusicas, y dado la existencia de diversos tipos de
cáncer de mama sensibles a estrógenos (positivos a receptores de estrógenos),
encontramos un factor más por el cual la obesidad contribuye a la carcinogénesis
mamaria.
Por último y definitivamente no menos importante, se ha observado que la
expresión de leptina y de su receptor obRb, se puede inducir bajo condiciones de
hipoxia (condición presente en tumores sólidos en etapa temprana) a través de la
activación del gen de leptina mediante el HIF-1. Esto es posible debido a que en
la región promotora del gen leptina en humanos, se presentan 8 regiones, las
cuales son capaces de responder y anclar al factor HIF-1, lo que activa la
transcripción. No suficiente con que esto, HIF-1 es un factor de transcripción
que se ha visto involucrado en respuesta al estrés nutricional que se presenta
en tumores sólidos, lo cual fomenta la angiogénesis del tumor y con ello
facilita el acceso de células neoplásicas circulación periférica que desemboca
en metástasis. Se ha determinado también, que la leptina puede inducir la
angiogénesis mediante la activación de factores involucrados en dicho proceso,
como lo son la metaloproteinasa de matriz (2 y 9), el VEGF y supresión de la
apoptosis en estas células vía un mecanismo dependiente de bcl-2. (5, 9)
Relación con otros receptores
En mujeres post-menopáusicas, el tejido adiposo es la única fuente de producción
de estrógenos. Dado que la actividad de la aromatasa y la síntesis de
androstenediona esta incrementada en este tejido, los niveles de estrógenos
resultantes en mujeres obesas están marcadamente elevados, además de que los
estrógenos sintetizados son rápidamente transformados en estradiol en la
circulación. Dicho estradiol presenta una mayor actividad biológica debido a la
deficiente unión de este con su proteína transportadora (SHGB), dejando libre al
estradiol de ejercer su efecto biológico con mayor potencia en diversas células
blanco. La importancia de esta sobre-exposición de estrógenos en mujeres post-menopáusicas
con obesidad radica en la existencia de una correlación positiva entre la
expresión por un lado; de los receptores de leptina obRb y estrógenos (RE), y
por el otro, del receptor de progesterona (RP) y leptina obRa.
Diversos estudios indican que la leptina incrementa la proliferación tanto de
células que son positivas a receptores de estrógenos como aquellas que no lo
son, ya que en un estudio realizado por Ray en el 2007(57) se encontró que la
leptina inducía la expresión de los receptores de estrógenos en líneas celulares
negativas a este receptor (MDA-MB-361, MDA-MB-231 y SK-BR-3) y dado que se ha
demostrado que los estrógenos inducen el crecimiento de las células neoplásicas,
además de incrementar la actividad de la enzima aromatasa, niveles elevados de
estrógenos aunados al estado de obesidad representan un factor muy importante en
la progresión y desarrollo de cáncer de mama dependiente de estrógenos en
mujeres post -menopáusicas.(5)
Otro estudio muy importante relacionado con los receptores hormonales y cáncer
mamario es el realizado por Hee Sun(13), en Corea, el cual concluyó que aquellos
pacientes que tenían niveles elevados de leptina pero negativos a la expresión
del receptor hormonal de estrógenos en las biopsias provenientes de pacientes
con cáncer mamario, presentaban un mayor porcentaje de sobrevivencia. La
condición leptina (+)/receptor hormonal (-) indicaba un buen pronóstico y una
buena respuesta a la quimioterapia, mientras que por el contrario, aquellos
pacientes con un perfil leptina (+)/ receptor hormonal (+) presentaban remisión
después de una cirugía de extirpación de alguna masa tumoral en alguno de los
senos y escasa respuesta a la quimioterapia. La importancia del perfil leptina
(+)/ receptor hormonal (-) radica en el hecho de que la leptina puede inducir la
expresión de receptores de hormonas (estrógenos), es decir, cambiar a un perfil
leptina (+)/ receptor hormonal (+) el cual está asociado con mal pronóstico. En
éste estudio también se demostró que existe muy poca correlación entre la
expresión de leptina y su receptor con variables como grado histológico de la
lesión (estado N y T en función de la American Joint Committee on Cancer),
estado de HER2, expresión de bcl-2 y de p53. Sin embargo una correlación
significativa fue hallada entre la expresión de leptina y su receptor con la
expresión del antígeno Ki-67, hecho que apoya la conceptualización de la leptina
como factor inductor de la carcinogénesis. (58) Este estudio refleja la relación
de los receptores de estrógenos con la leptina y sus receptores en el desarrollo
y progresión del cáncer mamario.
Entre otros receptores que tienen importancia en el estudio de leptina en la
carcinogénesis mamaria se encuentra a los receptores de adiponectina, cuya
hormona también es producida en el tejido adiposo, y que tiene un efecto
contrario a la leptina en las células neoplásicas de mama, inhibiendo su
crecimiento y fomentando su apoptosis, por lo que la expresión de este receptor
en las células neoplásicas de tejido mamario es de suma importancia al regular
los efectos que tiene la leptina en estas células.
Con lo antes mencionado se puede establecer que la leptina cumple un papel
fundamental en la patogénesis y progresión del cáncer mamario. Así como la falla
o poca respuesta ante terapias dirigidas en contra de los receptores de
estrógenos, donde fármacos como el tamoxifeno o inhibidores de aromatasa, tienen
como objetivo degradar rápidamente a estos receptores, pero que son bloqueados a
nivel citoplasmático por la leptina resultando en un estado de resistencia hacia
este tipo de tratamientos.
Interacción con otras hormonas y factores de crecimiento
Actualmente la evidencia existente a través de estudios realizados demuestra que
la leptina puede interactuar con los receptores tirosina-cinasa ErbB. Existen 4
miembros de estos receptores (EGFR-ErbB-1, ErbB-2 (HER2/Neu), ErbB-3 (HER3) y
ErbB-4 (HER4). En el caso de los tumores mamarios, pacientes HER2/neu positivas,
tienen peor pronóstico que las HER2 negativas (59). El gen HER2/neu está
implicado en el 20 a 25% de todos los carcinomas mamarios y está asociado con un
fenotipo de tumor agresivo y de mal pronóstico.(27) El tratamiento dirigido
hacia estas moléculas es con anticuerpos monoclonales como el Trastuzumab que va
dirigido específicamente hacia el receptor HER2. Bartsh,(60) en un estudio muy
reciente, demuestra que en pacientes con cáncer mamario metastásico a cerebro,
el empleo de Trastuzumab más Lapatinib (inhibidor de tirosina cinasas), aumenta
el porcentaje de sobrevida a la enfermedad en pacientes sometidas a este régimen
terapéutico. De igual manera, el uso de terapia convencional más el Trastuzumab
mejora considerablemente el pronóstico de las pacientes inhibiendo la
posibilidad de metástasis. Por otro lado, la co-expresión de HER2 y el sistema
leptina/obR podría contribuir a aumentar la actividad de HER2 y reducir la
sensibilidad a tratamientos anti HER2(61). La transforforilación de ErbB-2 por
los receptores obRa y obRb de leptina, activa las vías de señalización PI3K/AKT
y MAPK las cuales promueven fenómenos de proliferación y sobrevivencia de las
células neoplásicas de mama y aunque el mecanismo de interacción entre estos
receptores no ha sido elucidado del todo, se ha propuesto que la fosforilación
de ErbB lleva a cabo subsecuentemente a una activación de JAK2 por parte de los
receptores de leptina obR.(57, 61)
Otra hormona que se ha relacionado con la actividad de la leptina, es la
adiponectina, la cual se encarga de antagonizar los efectos de la primera en las
células neoplásicas del tejido mamario. Las concentraciones de adiponectina
disminuyen en personas obesas y existe una correlación entre obesidad y bajos
niveles de adiponectina. Recientemente, en algunos trabajos realizados por Jardé,
se encontró que la adiponectina inhibe el crecimiento de células de carcinoma
mamario, de la línea celular MCF-7 además de inducir la expresión de receptores
(AdipoR1 y AdipoR2) para esta hormona y disminuir la expresión tanto de leptina
como de sus receptores. Entre los receptores de adiponectina, se encontró que el
AdipoR2 se expresa en mayor cantidad que AdipoR1 y también se demostró la
diferencia significativa entre la expresión de AdipoR2 en células neoplásicas y
tejido adyacente normal al tumor. Estos estudios demuestran que la leptina tiene
un efecto pro-carcinogénico y la adiponectina anticancerígeno.(54, 62)
Conclusiones
La creciente incidencia de pacientes con cáncer de mama, así como el aumento de
los índices de obesidad y la relación existente entre ambas, se ha convertido en
un total desafío para la salud de la mujer Mexicana, por lo que el estudio y
elucidación de los mecanismos moleculares por los cuales la leptina induce el
desarrollo de las células neoplásicas permitirá encontrar nuevos blancos
terapéuticos contra el desarrollo y progresión de esta neoplasia.
Los niveles elevados de leptina en personas obesas son la causa por la que este
padecimiento resulta de gran importancia en el desarrollo y progresión del
cáncer de mama en mujeres post-menopáusicas, no sólo por la acción directa de la
leptina en las células neoplásicas a través de la vía de señalización
JAK2/STAT3, sino también por la inducción de la expresión de otros factores que
resultan ser de riesgo en la carcinogénesis del tejido mamario, como lo son los
receptores de estrógenos y activación de receptores ErbB. Estos hallazgos tienen
un gran impacto, que se deberían considerar en la práctica clínica, diagnóstico
y tratamiento del carcinoma mamario. La figura 4 resume lo antes mencionado.
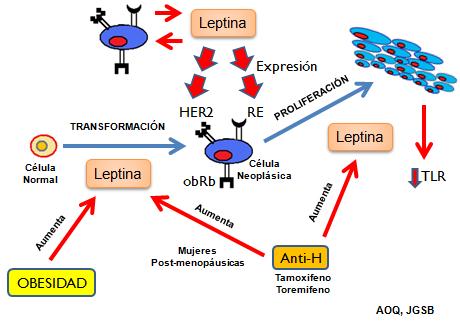
Figura 4. Importancia clínica de la Función de la Leptina en el desarrollo de
las células neoplásicas de cáncer de mama. TLR: tiempo libre de remisión, el
cual disminuye conforme aumenta los niveles de ARNm de leptina en las células
neoplásicas. (56) En la parte superior representa la idea de la relación
autocrina existente en cuanto a la producción y sitio de acción de la leptina en
las células neoplásicas.
Bibliografía
1. Yin N, Wang D, Zhang H, Yi X, Sun X, Shi B, et al. Molecular mechanisms
involved in the growth stimulation of breast cancer cells by leptin. Cancer Res.
2004;64(16):5870-5. Epub 2004/08/18.
2. Knaul FM, Nigenda G, Lozano R, Arreola-Ornelas H, Langer A, Frenk J. [Breast
cancer in Mexico: an urgent priority]. Salud Publica Mex. 2009;51 Suppl
2:s335-44. Epub 2010/01/09. Cancer de mama en Mexico: una prioridad apremiante.
3. Del Socorro Romero-Figueroa M, Santillan-Arreygue L, Miranda-Garcia M, Del
Pilar Torres-Arreola L, Perez-Espejel IM, Duarte-Mote J, et al. [Epidemiological
pattern of breast cancer mortality in Mexico State]. Rev Med Inst Mex Seguro Soc.
2010;48(3):253-8. Epub 2011/01/05. Patron epidemiologico de la mortalidad por
cancer de mama en el Estado de Mexico.
4. Villaseñor AD. La Obesidad en México. Salud Pública. 2011;Sect. Indicadores.
5. Garofalo C, Surmacz E. Leptin and cancer. J Cell Physiol. 2006;207(1):12-22.
Epub 2005/08/20.
6. Knaul FM, Nigenda G, Lozano R, Arreola-Ornelas H, Langer A, Frenk J. Cáncer
de mama en México: una prioridad apremiante. salud pública de méxico. 2009;51(supl
2).
7. Cárdenas Sanchez J. EVA, Maafs Molina E., Poitevin Chacón A. Consenso
Nacional sobre diagnóstico y tratamiento del cáncer mamario. México: Masson
Doyma México, S.A.; 2011 2011.
8. Salud Sd. NOM-041-SSA2-2011 Norma Oficial Mexicana para la prevención,
diagnóstico, tratamiento, control y vigilancia epidemiológica del cáncer de mama
Diario Oficial de la Federación 2011.
9. Cirillo D, Rachiglio AM, la Montagna R, Giordano A, Normanno N. Leptin
signaling in breast cancer: an overview. J Cell Biochem. 2008;105(4):956-64.
Epub 2008/09/30.
10. Eugenia FVVM. Señalización de la Leptina. Revista de Educación Bioquímica;
2006. p. 50-4.
11. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals.
Nature. 1998;395(6704):763-70. Epub 1998/10/31.
12. Hu X, Juneja SC, Maihle NJ, Cleary MP. Leptin--a growth factor in normal and
malignant breast cells and for normal mammary gland development. J Natl Cancer
Inst. 2002;94(22):1704-11. Epub 2002/11/21.
13. Kim HS. Leptin and leptin receptor expression in breast cancer. Cancer Res
Treat. 2009;41(3):155-63. Epub 2009/10/08.
14. Schwartz MW, Prigeon RL, Kahn SE, Nicolson M, Moore J, Morawiecki A, et al.
Evidence that plasma leptin and insulin levels are associated with body
adiposity via different mechanisms. Diabetes Care. 1997;20(9):1476-81. Epub
1997/09/01.
15. Havel PJ, Kasim-Karakas S, Mueller W, Johnson PR, Gingerich RL, Stern JS.
Relationship of plasma leptin to plasma insulin and adiposity in normal weight
and overweight women: effects of dietary fat content and sustained weight loss.
J Clin Endocrinol Metab. 1996;81(12):4406-13. Epub 1996/12/01.
16. Vadacca M, Margiotta DP, Navarini L, Afeltra A. Leptin in immuno-rheumatological
diseases. Cell Mol Immunol. China2011. p. 203-12.
17. Gonzalez-Perez RR, Xu Y, Guo S, Watters A, Zhou W, Leibovich SJ. Leptin
upregulates VEGF in breast cancer via canonic and non-canonical signalling
pathways and NFkappaB/HIF-1alpha activation. Cell Signal. England: 2010 Elsevier
Inc; 2010. p. 1350-62.
18. Chen DC, Chung YF, Yeh YT, Chaung HC, Kuo FC, Fu OY, et al. Serum
adiponectin and leptin levels in Taiwanese breast cancer patients. Cancer Lett.
2006;237(1):109-14. Epub 2005/07/16.
19. Mantzoros CS, Magkos F, Brinkoetter M, Sienkiewicz E, Dardeno TA, Kim SY, et
al. Leptin in human physiology and pathophysiology. American Journal of
Physiology-Endocrinology And Metabolism. 2011;301(4):E567-E84.
20. Myers MG, Jr. Leptin receptor signaling and the regulation of mammalian
physiology. Recent Prog Horm Res. 2004;59:287-304. Epub 2004/01/30.
21. Li C, Friedman JM. Leptin receptor activation of SH2 domain containing
protein tyrosine phosphatase 2 modulates Ob receptor signal transduction.
Proceedings of the National Academy of Sciences of the United States of America.
1999;96(17):9677-82. Epub 1999/08/18.
22. Banks AS, Davis SM, Bates SH, Myers MG, Jr. Activation of downstream signals
by the long form of the leptin receptor. J Biol Chem. United States2000. p.
14563-72.
23. Carpenter LR, Farruggella TJ, Symes A, Karow ML, Yancopoulos GD, Stahl N.
Enhancing leptin response by preventing SH2-containing phosphatase 2 interaction
with Ob receptor. Proceedings of the National Academy of Sciences of the United
States of America. 1998;95(11):6061-6. Epub 1998/05/30.
24. Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, et al. SOCS3
mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem.
United States2000. p. 40649-57.
25. Hekerman P, Zeidler J, Bamberg-Lemper S, Knobelspies H, Lavens D, Tavernier
J, et al. Pleiotropy of leptin receptor signalling is defined by distinct roles
of the intracellular tyrosines. FEBS J. England2005. p. 109-19.
26. Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Munzberg H, Myers MG,
Jr. The long form of the leptin receptor regulates STAT5 and ribosomal protein
S6 via alternate mechanisms. J Biol Chem. United States2007. p. 31019-27.
27. Guo S, Liu M, Wang G, Torroella-Kouri M, Gonzalez-Perez RR. Oncogenic role
and therapeutic target of leptin signaling in breast cancer and cancer stem
cells. Biochim Biophys Acta. Netherlands: A 2012 Elsevier B.V; 2012. p. 207-22.
28. Procaccini C, Lourenco EV, Matarese G, La Cava A. Leptin signaling: A key
pathway in immune responses. Current signal transduction therapy.
2009;4(1):22-30. Epub 2009/09/24.
29. Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG, Jr. Regulation of
Jak kinases by intracellular leptin receptor sequences. J Biol Chem. United
States2002. p. 41547-55.
30. Shen J, Sakaida I, Uchida K, Terai S, Okita K. Leptin enhances TNF-alpha
production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sci.
England2005. p. 1502-15.
31. Donato J, Jr., Frazao R, Elias CF. The PI3K signaling pathway mediates the
biological effects of leptin. Arquivos brasileiros de endocrinologia e
metabologia. 2010;54(7):591-602. Epub 2010/11/19.
32. Myers MG, Jr., Backer JM, Sun XJ, Shoelson S, Hu P, Schlessinger J, et al.
IRS-1 activates phosphatidylinositol 3'-kinase by associating with src homology
2 domains of p85. Proceedings of the National Academy of Sciences of the United
States of America. 1992;89(21):10350-4. Epub 1992/11/11.
33. Huang XF, Chen JZ. Obesity, the PI3K/Akt signal pathway and colon cancer.
Obes Rev. England2009. p. 610-6.
34. Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, et al. Concomitant
activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated
promotion of invasion and migration of hepatocellular carcinoma cells. Cancer
Res. United States2007. p. 2497-507.
35. Johnston A, Arnadottir S, Gudjonsson JE, Aphale A, Sigmarsdottir AA,
Gunnarsson SI, et al. Obesity in psoriasis: leptin and resistin as mediators of
cutaneous inflammation. Br J Dermatol. England2008. p. 342-50.
36. Kumar S, Kishimoto H, Chua HL, Badve S, Miller KD, Bigsby RM, et al.
Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model
of breast cancer. Am J Pathol. United States2003. p. 2531-41.
37. Maedler K, Sergeev P, Ehses JA, Mathe Z, Bosco D, Berney T, et al. Leptin
modulates beta cell expression of IL-1 receptor antagonist and release of
IL-1beta in human islets. Proceedings of the National Academy of Sciences of the
United States of America. United States2004. p. 8138-43.
38. Carino C, Olawaiye AB, Cherfils S, Serikawa T, Lynch MP, Rueda BR, et al.
Leptin regulation of proangiogenic molecules in benign and cancerous endometrial
cells. International journal of cancer Journal international du cancer.
2008;123(12):2782-90. Epub 2008/09/19.
39. Zhou W, Guo S, Gonzalez-Perez RR. Leptin pro-angiogenic signature in breast
cancer is linked to IL-1 signalling. Br J Cancer. England2011. p. 128-37.
40. Garofalo C, Koda M, Cascio S, Sulkowska M, Kanczuga-Koda L, Golaszewska J,
et al. Increased expression of leptin and the leptin receptor as a marker of
breast cancer progression: possible role of obesity-related stimuli. Clin Cancer
Res. 2006;12(5):1447-53. Epub 2006/03/15.
41. Smirnova OV, Ostroukhova TY, Bogorad RL. JAK-STAT pathway in carcinogenesis:
is it relevant to cholangiocarcinoma progression? World J Gastroenterol.
2007;13(48):6478-91. Epub 2007/12/29.
42. Behera R, Kumar V, Lohite K, Karnik S, Kundu GC. Activation of JAK2/STAT3
signaling by osteopontin promotes tumor growth in human breast cancer cells.
Carcinogenesis. 2010;31(2):192-200. Epub 2009/11/21.
43. Garofalo C, Surmacz E. Leptin and cancer. Journal of cellular physiology.
2006;207(1):12-22.
44. Tomita N. BCL2 and MYC dual-hit lymphoma/leukemia. J Clin Exp Hematop.
2011;51(1):7-12. Epub 2011/06/02.
45. Das SN, Khare P, Singh MK, Sharma SC. Correlation of cyclin D1 expression
with aggressive DNA pattern in patients with tobacco-related intraoral squamous
cell carcinoma. Indian J Med Res. 2011;133(4):381-6. Epub 2011/05/04.
46. Gareau C, Fournier MJ, Filion C, Coudert L, Martel D, Labelle Y, et al.
p21(WAF1/CIP1) upregulation through the stress granule-associated protein CUGBP1
confers resistance to bortezomib-mediated apoptosis. PLoS One. 2011;6(5):e20254.
Epub 2011/06/04.
47. Silvestre DC, Gil GA, Tomasini N, Bussolino DF, Caputto BL. Growth of
peripheral and central nervous system tumors is supported by cytoplasmic c-Fos
in humans and mice. PLoS One. 2010;5(3):e9544. Epub 2010/03/09.
48. Naderi A, Liu J, Hughes-Davies L. BEX2 has a functional interplay with
c-Jun/JNK and p65/RelA in breast cancer. Mol Cancer. 2010;9:111. Epub
2010/05/21.
49. Farras R, Baldin V, Gallach S, Acquaviva C, Bossis G, Jariel-Encontre I, et
al. JunB breakdown in mid-/late G2 is required for down-regulation of cyclin A2
levels and proper mitosis. Mol Cell Biol. 2008;28(12):4173-87. Epub 2008/04/09.
50. El-Asrar AM, Missotten L, Geboes K. Expression of high-mobility groups
box-1/receptor for advanced glycation end products/osteopontin/early growth
response-1 pathway in proliferative vitreoretinal epiretinal membranes. Mol Vis.
2011;17:508-18. Epub 2011/03/03.
51. V GG, MA GM, A BM. Expresión de bcl-2, ki-67 y caspasa-3
en lesiones cancerosas de la mucosa oral.
Resultados preliminares. Avances en Odontoestomatología. 2006;22(5):263-9.
52. Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA.
Identification of a genetic signature of activated signal transducer and
activator of transcription 3 in human tumors. Cancer Res. 2005;65(12):5054-62.
Epub 2005/06/17.
53. Perrier S, Caldefie-Chezet F, Vasson MP. IL-1 family in breast cancer:
potential interplay with leptin and other adipocytokines. FEBS Lett.
Netherlands2009. p. 259-65.
54. Jarde T, Caldefie-Chezet F, Goncalves-Mendes N, Mishellany F, Buechler C,
Penault-Llorca F, et al. Involvement of adiponectin and leptin in breast cancer:
clinical and in vitro studies. Endocr Relat Cancer. 2009;16(4):1197-210. Epub
2009/08/08.
55. Ishikawa M, Kitayama J, Nagawa H. Enhanced expression of leptin and leptin
receptor (OB-R) in human breast cancer. Clin Cancer Res. 2004;10(13):4325-31.
Epub 2004/07/09.
56. Revillion F, Charlier M, Lhotellier V, Hornez L, Giard S, Baranzelli MC, et
al. Messenger RNA expression of leptin and leptin receptors and their prognostic
value in 322 human primary breast cancers. Clin Cancer Res. 2006;12(7 Pt
1):2088-94. Epub 2006/04/13.
57. Ray A, Nkhata KJ, Cleary MP. Effects of leptin on human breast cancer cell
lines in relationship to estrogen receptor and HER2 status. Int J Oncol.
2007;30(6):1499-509. Epub 2007/05/10.
58. Rakovitch E, Nofech-Mozes S, Hanna W, Narod S, Thiruchelvam D, Saskin R, et
al. HER2/neu and Ki-67 expression predict non-invasive recurrence following
breast-conserving therapy for ductal carcinoma in situ. Br J Cancer.
England2012. p. 1160-5.
59. Cortes J, Saura C, Bellet M, Munoz-Couselo E, Ramirez-Merino N, Calvo V, et
al. HER2 and hormone receptor-positive breast cancer--blocking the right target.
Nat Rev Clin Oncol. England2011. p. 307-11.
60. Bartsch R, Berghoff A, Pluschnig U, Bago-Horvath Z, Dubsky P, Rottenfusser
A, et al. Impact of anti-HER2 therapy on overall survival in HER2-overexpressing
breast cancer patients with brain metastases. Br J Cancer. England2012. p.
25-31.
61. Fiorio E, Mercanti A, Terrasi M, Micciolo R, Remo A, Auriemma A, et al.
Leptin/HER2 crosstalk in breast cancer: in vitro study and preliminary in vivo
analysis. BMC Cancer. 2008;8:305. Epub 2008/10/24.
62. Jardé T, Perrier S, Vasson MP, Caldefie-Chézet F. Molecular mechanisms of
leptin and adiponectin in breast cancer. European Journal of Cancer.
2011;47(1):33-43.