ISSN 1666-7948
www.quimicaviva.qb.fcen.uba.ar
“Del Vuelo de las Proteínas y de como lograrlo”
(Espectrometría de masa UV-MALDI)
por la Dra. Rosa Erra-Balsells
CIHIDECAR-CONICET, Departamento de Química Orgánica, Facultad de Ciencias Exactas y Naturales, UBA
Recibido 10 de marzo de 2004/ Aceptado 13 de abril de 2004

El Premio Nobel de Química 2002 fue compartido por tres investigadores, dos especialistas en Espectrometría de Masa (MS) y uno en Resonanica Magnética Nuclear (NMR) dado que gracias a sus contribuciones estas técnicas pueden usarse para el análisis de macromoléculas biológicas y en especial para el de proteínas (Fig. 1).

Figura 1. Premio Nobel de Química 2002
(www.nobel.se/chemistry/
laureates/ 2002)
Básicamente, en un espectrómetro de masa se convierte a las macromoléculas (analito) en iones gaseosos y éstos son diferenciados por el analizador según su relación m/z (z, carga del ion; m, masa molar o peso molecular del ion), obteniéndose así información de su peso molecular (Fig. 2) [1].
¿Por qué es importante el análisis de proteínas por Espectrometría de Masa?
Para entender el mecanismo por el cual actúa una proteína in vivo se requiere una completa caracterización de la misma en lo que se refiere a estructura primaria, peso molecular y estructura tridimensional o forma de la proteína. La proteína desde su síntesis y durante su traslado al punto de acción puede sufrir cambios estructurales (modificaciones post-traduccionales) los que modifican su estructura primaria y como consecuencia su peso molecular. La proteína puede ajercer su función específica no como una unidad monomérica tal cual fue sintetizada, sino como agregados no covalentes del tipo dímero, trímero, tetrámero, etc. Para monitorear todas estas posibilidades se requieren técnicas analíticas precisas, de alta sensibilidad y en lo posible rápidas.
Hasta la década del 90, la determinación del peso molecular y la estructura primaria de las proteínas requería el uso combinado de las técnicas electroforéticas, cromatográficas y de la degradación de Edman. Este análisis en su conjunto es muy laborioso y requiere una considerable cantidad de tiempo. A mediados de la década del 90, el reemplazo de estas técnicas por la espectrometría de masa ha dado lugar al área de investigación conocida como “Proteomics” [2, 3].
De la misma manera, de la aplicación de las espectrometría de masa al análisis de otras familias de macromoléculas biológicas tales como hidratos de carbono, lípidos y metabolitos secundarios han surgido nuevas áreas de investigación denominadas “Glycomics”, “Lipidics” y “Metabolomics” [3, 4].
Espectrometría de masa
Todos los espectrómetros de masa constan de tres partes esenciales. La primera es la “fuente”, en la que se generan los iones gaseosos del analito (volatilización/ionización). La segunda es el “analizador de masas” ya que diferencia a los iones gaseosos según su relación m/z. La tercera es el “detector” el cual recibe a los iones diferenciados por el analizador (Fig. 2).
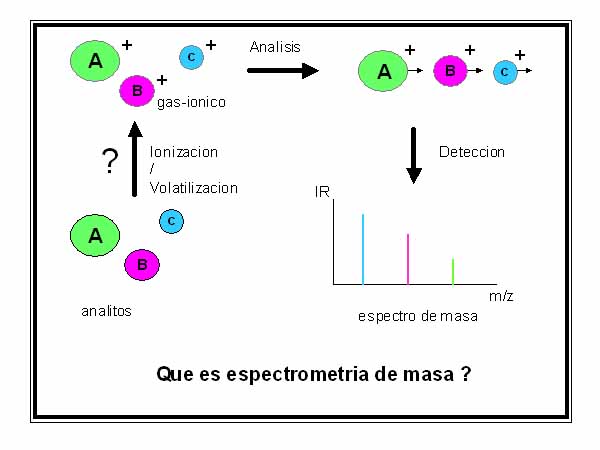
Figura 2. Procesos que ocurren dentro de un espectrómetro
de masa
Un cuarto elemento fundamental del equipo son las bombas de vacío, que regulan los valores absolutos y el gradiente de vacío entre las tres partes mencionadas. A todo estos se agrega en la actualidad computadoras de alta capacidad operativa para la realización del experimento en sí, almacenamiento y manejo de datos.
Los avances tecnológicos y la investigación básica han contribuído al desarrollo en muchos casos en forma independiente de las tres partes mencionadas. Existen en la actualidad diversos tipos de “fuentes” basadas en fenómenos físicos y físico-químicos de volatilización y ionización completamente diferentes. Existen también diferentes tipos de analizadores. De todas las combinaciones posibles surge la variedad de espectrómetros de masa que hoy se ofrecen en el mercado.
Las preguntas naturales surgen:
¿Porqué tantas opciones?
¿Cuál de las opciones actuales es útil para bio-moléculas?
¿Por qué razón Koichi Tanaka compartió el Premio Nobel de Química 2002 con John B. Fenn (Fig. 1), ambos por sus contribuciones al desarrollo de espectrometrías de masa útiles para el análisis de proteínas y bio-macromoléculas en general?
En realidad contribuyeron independientemente sólo al desarrollo de dos nuevas “fuentes” de ionización (UV-MALDI y ESI). ¿Es eso tan importante?
¿Qué es UV-MALDI?
¿Qué es ESI ?
Un poco de historia
El uso de la espectrometría de masa (MS) como técnica analítica creció en forma exponencial desde que J. J. Thompson describiera en 1910 experimentos en los que iones gaseosos producidos por bombardeo electrónico de especies gaseosas neutras eran separados según el valor de su relación m/z (m, masa del ion en unidades de masa atómica de daltons (Da)) [5]. Si bien este descubrimiento llevó a experimentos clave para la caracterización de isótopos estables (descubrimiento del deuterio, Premio Nobel de Química 1934, Harold Urey) y radionucleídos, las aplicaciones de esta técnica se vieron limitadas a compuestos (analitos) de bajo peso molecular, termoestables y fácilmente volatilizables. Durante muchos años, la generación de moléculas gaseosas iónicas pasaba por dos etapas, la primera de volatilización del analito haciendo uso de calor y la segunda de ionización del analito gaseoso neutro mediante el bombardeo del mismo con especies que lo ionizaran (Fig. 2). De esta manera a la técnica de ionización por bombardeo con una haz de electrones acelerados (ionización por impacto de electrones, EI) [6, 7] siguió el bombardeo con un haz de moléculas gaseosas ionizantes (ionización química, CI) [6, 7] y también la exposición del analito gaseoso a la acción de un fuerte campo eléctrico que produce su ionización por efecto túnel (ionización por campo, FI) [7].
Pese a la limitación de que la técnica era útil solo para analitos termoestables, la espectrometria de masa progresó constantemente desde su invención casi cien años atrás. El fuerte desarrollo se debió tanto a los avances tecnológicos por un lado (mejoras instrumentales: desarrollo de nuevos analizadores de iones gaseosos; desarrollo de nuevos detectores; aumento de la sensibilidad y rapidez de los experimentos; procesamiento de datos; banco de datos; etc) como al avance en el conocimiento del comportamiento de los iones moleculares gaseosos (cationes, cationes radicales, aniones, aniones radicales; su estabilidad y sus formas de descomposición o fragmentación [8, 9]). Como ya se señaló, todos los espectrómetros de masa tiene en común que miden la relación m/z de iones gaseosos haciendo uso de campos eléctricos y/o magnéticos (Fig. 2). La exacta medida de este valor para las moléculas gaseosas ionizadas del analito (ion molecular) dará información sobre su masa molar (peso molecular). Si además se induce la fragmentación del ion molecular, cada fragmento iónico será detectado con su propio valor de m/z. El conjunto de fragmentos formados es característico de cada analito y depende de su estructura química (“huella dactilar”). Por lo tanto, del análisis de los fragmentos obtenidos se obtiene información sobre la estructura química del analito en cuestión [6-9].
El uso de la espectrometría de masa (MS) apunta siempre a los dos aspectos mencionados: (i) determinación de masas molares (pesos molecualres) y (ii) determinación de estructura química (Fig. 3).

Figura 3. Utilidad de la espectrometría de masa
La imposibilidad de aplicación de la técnica a analitos termolábiles limitó durante muchos años el posible uso de la misma en el campo de las biomoléculas. Se requería, previo al análisis, la preparación de derivados termoestables. La engorrosa preparación de los mismos (por ejemplo en el campo de los hidratos de carbono se preparaban derivados per-metilados o per-acetilados), hacía que muy pocos se vieran atraídos por el su uso de esta técnica. Además, se requerían cantidades significativas de muestra para su manipuleo químico previo al análisis por MS [6-9].
En la decada del 70 se describen los primeros experimentos exitosos donde moléculas termolábiles eran transformadas sin descomposición alguna y en un único paso a iones gaseosos. Estas técnicas de volatilización/ionización se denominan “técnicas de desorción” (Fig. 4). En estos casos al analito no volátil convenientemente depositado sobre una superficie metálica (porta muestra) en alto vacío, es bombardeado con un haz de átomos neutros acelerados (He: Xe; “fast atom bombardment”, FAB) [7, 10], o con un haz de átomos iónicos acelerados (He+; Xe+; “static secondary ion MS, SIMS o liquid secondary ion MS”, LSIMS) [7, 11], o es expuesto a la acción de un fuerte campo eléctrico que induce su volatilización/ionización por efecto túnel (field desorption, FD) [7, 12], o es expuesto a la acción de un plasma (plasma desorption MS; PD) [7] o a la acción de un láser (laser desortion ionization, LD) [7, 13]. Si bien estos métodos de ionización ampliaron el uso de la técnica a moléculas termolábiles y a macromoléculas ya sea sintéticas o biomacromoléculas, prácticamente su límite de aplicación es para analitos de peso molecular por debajo de 700-800 Da. En ese momento, una de las revistas más importantes de espectrometría de masa todavía se llamaba J. Org. Mass Spectrom. [14]. Los prinicipales usuarios de la técnica seguían siendo los químico orgánicos que trabajaban con analitos termoestables de peso moelcular bajo (< 800 Da).
El progreso había sido notable, se median pesos moleculares cercanos a 1kDa con alta resolución con aproximación en el cuarto decimal pero... “el análisis de las grandes macromoléculas como moléculas intactas gaseosas ionizadas seguia siendo un deseo, un sueño”.
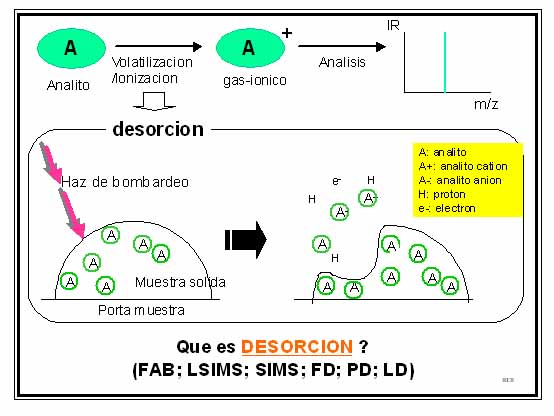
Figura 4.
Volatilización/ionización de analitos por desorción.
El sueño se hizo
realidad…”Las macromoléculas vuelan”
En particular los intentos de usar un láser ultravioleta como fuente de energía para la provocar desorción se remonta a la década del 60. Se lo usó primeramente para realizar determinaciones de composición elemental y luego para la desorción de biomoléculas pequeñas, siendo el mayor problema experimental que el analito debía absorber los fotones emitidos por la fuente láser ultravioleta, generando como especie primaria un real estado electrónico excitado. A partir del mismo muchos analitos reaccionan fotoquímicamente o en otros fotoquímicamente estables el exceso de energía vibracional residual en la molécula desorbida induce su fragmentación inmediata. Cualquiera de las dos posibilidades enunciadas, en las que se usaba el láser Nd:YAG (lem 266 nm), imposibilitan el análisis del ion molecular gaseoso intacto como tal a fin de obtener informacion del valor de su m/z y peso molecular [15].
La idea de usar una fuente láser pulsada ya sea en la región UV-visible o en la región infrarroja (IR) para la desorción ya existía. Todos los laboratorios se afanaban en ese momento en realizar experimentos con el segundo tipo de fuente láser, ya que los órdenes de energía que se requieren para “deformar” la molécula y así “aislarla” de su entorno en el medio sólido y permitir su “desorción” los provee un Láser IR.
Sin embargo, el “hecho mágico” producto quizás en parte de la casualidad se comunica en un congreso en el año 1987 [16]. El Ing. Koichi Tanaka (Premio Nobel de Química 2002), quien realizó el experimento, en su exposición con motivo de la recepción del galardón [17], aclara que el no es químico y que su falta de prejuicio sobre la “imposibilidad práctica” de volatilizar macromoléculas termolábiles por calentamiento más ciertos errores al preparar las muestra lo llevó a usar como analitos polímeros (polietilenglicoles) y proteínas a las que mezcló con polvo metálico de una granulación especial (ultra fine metal powder (patente japonesa) UFMP; diámetro, decena de nm; de aplicación en metalurgia por la eficiente transmisión de calor [17]) usando propilenglicol en lugar de acetona, como medio para generar una mezcla sólida de analito+polvo metálico. La idea de usar UFMP mezclado con el analito a ser bombardeado por el láser, era aprovechar todo el “calor rápido” generado por el láser para aumentar la velocidad de evaporación del analito de modo que fuera mucho mayor que la de su descomposición térmica [17]. Al transferir la suspensión constituída por macromolecula + polvo metálico en acetona sobre la superficie de portamuestras introducido en la cámara de ionización en alto vacio, la acetona se evaporaba ...y en realidad generaba sobre el porta muestra una muestra sólida heterogénea constituída por polvo metalico “embebiendo” al analito en estado sólido (como pasas en un budín). Cuando por error usó propilenglicol, en lugar de tirar la preparación pastosa obtenida sobre el porta muestra, lo introdujo en la cámara de ioniación, en alto vacío, pensando que el propilenglicol se evaporaría durante el experimento. Esta suspensión pastosa con distribución más homogénea del analito y del polvo metálico al ser bombardeada por el láser ultravioleta de nitrógeno (lem 337 nm) produjo el resultado “mágico”, deseado y largamente esperado, de generar iones moleculares intactos gaseosos del analito macromolecular en cuestión (proteínas y polímeros sintéticos) [18]. En las presentaciones en congresos y publicaciones del período 1987-1988 [16-18], Tanaka mostró que era posible con un láser de N2 generar los iones moleculares intactos de quimotripsinógeno (25.717 Da), carboxipeptidasa-A (34.472 Da), citocromo c (12.384 Da) y polietilenglicol (PEG 2000 Da). El había logrado el resultado deseado usando, entre otras cosas, un láser utravioleta de baja energía (Laser de N2, lem 337nm), cuya radiación no era absorbida ni por las proteínas ni por el PEG, y esto era completamente inesperado. Simultáneamente en el mismo año (1998, [19]), Karas y Hillenkamp describen la detección del ion gaseoso intacto de proteínas. A diferencia del experimento de Tanaka, ellos depositan sobre el porta muestra una mezcla sólida donde la proteína (analito) sigue siendo el componente minoritario y está disperso o “embebido” como “pasas en el budin” en el componente mayoritario (matriz). Ahora la matriz es simplemente un “pequeña” molécula orgánica fotosensible ya que debe absorber la radiación provista por el Láser (337 nm), radiación a la cual la proteína es transparente (Fig. 5). Si bien este es el pricipio básico con el que se rige actualmente la preparación de muestras para UV-MALDI, estos autores en su primer trabajo usaron como matriz al ácido nicotínico y la fuente empleada fue un láser Nd:YAG (lem 266 nm). En sus condiciones experimentales tanto matriz con analito absorbían radiación. Sin embargo entre las múltiples señales obtenidas pudieron ver la correspondiente al ion molecular intacto de la proteína. El uso de matrices metálicas (soft laser desorption, SLD) prácticamente no ha encontrado hasta ahora mayor aplicación analítica. El uso de las matrices orgánicas fotosensibles ha dado lugar al desarrollo del método de volatilización/ionziación suave, que hoy se conoce como “ultraviolet matrix assisted laser desorption/ionization mass spectrometry” UV-MALDI. Sin embargo se considera que SLD es la idea madre en la que se basa UV-MALDI y otros métodos de desorción/ionización actualmente en desarrollo tales como SELDI (Surface Enhanced Laser Desorptio Ionization) y DIOS (Direct Ionisation on Silicon).
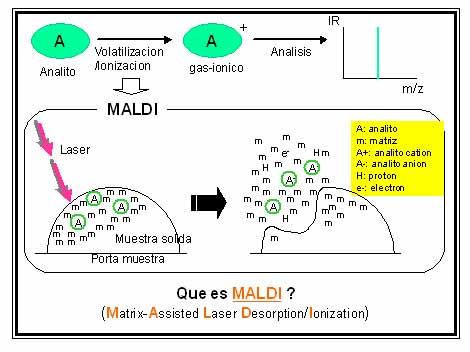
Figura 5.
Volatilización/ionización de analitos por MALDI.
Algunas características de
UV-MALDI-MS
Desde su implementación práctica a fines de la década del 80, el método de ionización UV-MALDI se ha usado con éxito para el análisis por espectrometría de masa
de bio-macromoléculas (ácidos nucléicos, nucleótidos, nucleósidos, proteínas, péptidos, lípidos, hidratos de carbono, compuestos glicoconjugados, etc) y polímeros sintéticos. Ha demostrado reunir una serie de ventajas, siendo unas de las más importantes la simplicidad espectral y el rango de valores de m/z en que es aplicable. Asi, para macromoléculas con masas moleculares de hasta 500 kDa controlando la condiciones experimentales (“potencia laser umbral”) se producen casi exclusivamente los iones moleculares monocargados (m/z con z=1) estables, sin inducirse su fragmentación (cada señal observada corresponde en principio al ion molecular de una especie química). Por lo tanto es posible analizar tanto compuestos puros como mezclas y cuantificar las segundas. En esta propiedad del UV-MALDI se basa no sólo el uso clásico como una espectrometría de masa para determinar pesos moleculares sino además su uso para el “mapeo de mezclas”, por ej. “mapeo de proteínas” o “mapeo de polipéptidos” muy usado para la caracterización de bio-sistemas (por ej., control de adulteración de muzzarella con leche de cabra y/o de vaca [20]) asi como para la obtención de “imágenes de tejidos” en el diagnóstico de enfermedades [21], o simplemente carcterizar la mezcla de péptidos obtenidos luego de la digestión con tripsina de un polipéptido o de una proteína, “huella peptídica” [2a] ). Gracias al desarrollo tecnológico actual y al conocimiento que se tiene desde los 60 del comportamiento promedio de moléculas gaseosas iónicas, pequeñas y medianas, según sea la estructura y ubicación de sus grupos funcionales (reglas de fragmentaciones y reordenamientos de compuestos orgánicos y biomoléculas pequeñas [6, 8, 9]) en la actualidad se pueden realizar experimentos donde no sólo se determinan pesos moleculares sino que además se induce la fragmentación específica de un ion pre-seleccionado (“precursor ion” o “father ion”) y analizando las señales del espectro que origina (estudiando su esquema de fragmentación) se obtiene información de su estructura (tipo de grupo funcionales, bloques estructurales, secuencia en que están unidos, etc) (Fig. 6). O sea que dos especies con la misma fórmula molecular (igual composición centesimal) y el mismo peso molecular pueden diferenciarse perfectamente por el esquema de fragmentacion de su ion molecular (ej.: los péptidos VGAHAGEYGAEALER y IGGHGAEYGAEALER tienen el mismo peso molecular (m/z) pero su esquema de fragmentación es diferente, [2a]) .
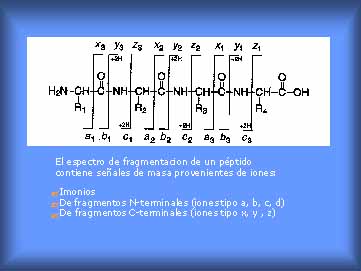
Figura 6. Fragmentaciones principales de péptidos.
Dado un método de generación de iones gaseosos, la sensibilidad, resolución y la exactitud con la que se determina el valor de la m/z depende fuertemente del tipo de analizador de iones empleado. En los casos que las masas molares de los iones se determina con aproximación en el cuarto o quinto decimal se habla de espectrometría de masa de alta resolución (HRMS). Para un valor de m/z asi determinado, es posible asociar una o unas pocas combinaciones de átomos constituyentes para su fórmula molecular. Cuando menor es el valor de m/z determinado por HRMS, menor es el número de combinaciones atómicas posibles. Si para un péptido se determina primero con alta exactitud (HRMS) su m/z y luego se realiza un experimento en el que se induce la fragmentación y para los fragmentos generados se determina con alta axactitud (HRMS) su m/z, es posible entrando a bancos de datos mediante programas especialmente desarrollados para esta familia de compuestos, obtener información sobre la posible estructura en cuanto a aminoácidos componentes y secuencia de los mismos en la cadena de los fragmentos y del péptido original. Otra variante es la tecnología denominada “a novo” por la cual el banco de datos genera (simula) los espectros con todas las fragmentaciones para cada uno de los polipéptidos posibles según el valor m/z obtenido experimentalmente para el polipéptido en estudio. Luego compara el espectro obtenido cuando se induce experimentalmente la fragmentación del polipéptido en cuestión con los obtenidos por simulación (Fig. 7).

Figura 7. Programas y bancos de datos de uso actual para el
análisis de proteínas por espectrometría de masa [ref. 2].
Pese a las limitaciones prácticas y condiciones de contorno que exige un uso confiable de estos programas y banco de datos, esta posibilidad ha revolucionado la analítica de las proteínas y polipéptidos, por ello se ha producido una verdadera explosión en el uso de la espectroscopía UV-MALDI MS. En la actualidad hablar del desarrollo de proyectos “proteomics” significa contar, entre otras, con esta herramienta analítica esencial (Fig. 8).
Las mismas posibilidades de uso se dan también para las demás familias de bio-macromoléculas. Sin embargo, no se cuenta aun con el apoyo de bancos de datos y programas que permitan, una vez obtenido los espectros de masa (ion molecular y luego fragmentación del mismo) una rápida y eficaz búsqueda de estructuras probables o una precisa simulación “a novo” de los diagramas de fragmentación teóricos (Figs. 8 y 9). Este último tipo de simulación también se ha intentado en el campo de los oligosacáridos, pero su utlidad es aun muy reducida (programa STAT) [22].
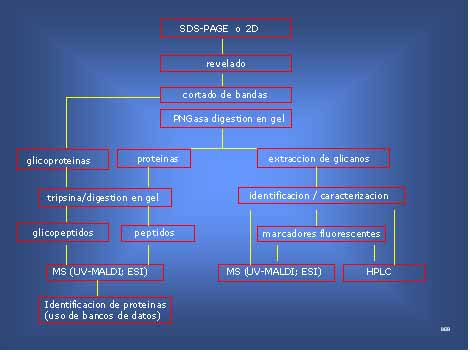
Figura 8. Combinación de técnicas para la caracterización de glicoproteínas haciendo uso de espectrometría de masa (MS)
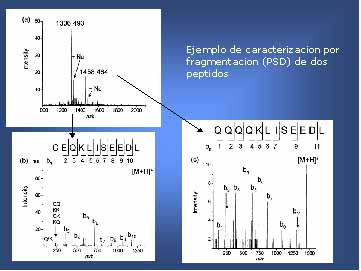
Figura 9. UV-MALDI-TOF MS análisis de un polipéptido
fragmentado en dos péptidos de peso molecular 1458,464 y 1306, 493 (detectados
como M+H+) (huella peptídica).
Espectros PSD (inferior izquierdo y derecho) de ambos péptidos.
Algunos detalles sobre equipamiento con UV-MALDI disponible
Si el espectrómetro de masa tiene solamente como analizador un “tiempo-de-vuelo” (TOF) se efectúan los experimentos primero en modo “lineal “ y “reflectrón” (mayor exactitud en m/z a expensas de menor sensibilidad en el segundo) para determinar el peso molecular (condiciones experimentales de no fragmentación) y luego, mediante un modo de operación que se denomina PSD (“post source decomposition”) se induce la fragmentación del ion seleccionado. Este sería un equipo UV-MALDI-TOF MS preparado para trabajar en el modo PSD [7]. No todos los equipos UV-MALDI-TOF MS tienen esta opción. La sensibilidad, resolución y exactitud varían según las marcas y los modelos. En la actualidad existen equipos más sofisticados donde al método de ionización UV-MALDI se lo combina con analizadores también únicos que permiten hacer los dos experimentos antes mencioandos (sin fragmentación y con fragmentación). Estos analizadores son la denomianda “trampa iónica” (IT) y el llamado “fourier transform-ion ciclotron resonance” (FT-ICR) y los equipos serían UV-MALDI-IT y UV-MALDI-FT-ICR [7]. La resolución, exactitud y sensibiliad son muy altas. También existen los equipos que tienen en un arreglo “Tandem” dos analizadores de iones y los experimentos realizados se los denomina “espectrometría de masa tandem” (por ej. MS/MS o MS2 ). El equipo clásico es el denominado UV-MALDI-Q-TOF que combina un cuadrupolo (Q) y el analizador TOF y también existe la combinación UV-MALDI-IT-TOF [7]. Todos estos equipos tienen aún en la actualidad la seria limitación de que permiten analizar iones gaseosos de m/z < 15 kDa. Sólo el primer tipo de equipo mencionado, UV-MALDI-TOF y los de nueva generación que han aparecido en el mercado poniendo en “tandem” dos analizadores TOF (UV-MALDI-TOF-TOF) no tendrían “teóricamente” límite superior en cuanto al valor de m/z máximo que pueden analizar y focalizar al detector. Si bien esto prácticamente es correcto en los primeros (se han detectado proteínas de m/z < 300 kDa), es aun prematuro afirmar lo mismo en los segundos.
Matrices y algunas limitaciones experimentales
Para los experimentos UV-MALDI MS se require para preparar la muestra y efectuar el análisis volúmenes de solución del fotosensibilizador (matriz) del orden del microlitro, siendo la relación de concentración entre el analito y el fotosensibilizador (matriz) del orden de 1:1000 a 1:100000 mol/mol. Independientemente de como se deposite las mezclas sólida sobre el portamuestra, la matriz generará siempre las señales mas intensas del espectro, especialmente en la región cercana al valore de m/z de su ion molecular. El papel de la matriz es activo durante el experimento ya al ser la responsable de la absorción de los fotones, induce su propia volatilización/ionización (desorción) y la del (o de los) analitos (Fig. 5).
La absorción de los fotones por parte de un compuesto orgánico es condición necesaria pero no suficiente para actuar como matriz UV-MALDI.
Sin embargo, uno de los serios problemas que tiene aun esta técnica es que no se conocen bien las condiciones que deben cumplir el par analito-matriz para que la desorción/ionización del analito, como unidad monomérica inalterada, sea eficaz. O sea que debe ser eficiente además de la volatilización de las moléculas aisladas del analito, su ionización ya sea por intercambio de electrones y/o de protones o por mantenimiento de una interacción de tipo donante-aceptor de electrones (orbitales de tipo n o de tipo p y orbitales vacantes) con unidades que tienen carga neta (ej. Na+, K+), durante la volatilización.
Pese a la difusión del uso de la espectrometría de masa UV-MALDI y a su indiscutible utilidad en el campo de las macromoléculas biológicas y sintéticas, la búsqueda de nuevas matrices y la comprensión de los procesos involucrados en la desorción/volatilización del analito “asistida” (fotosensibilizada) por la matriz sigue siendo aun tema de investigación y estudio [23].
Referencias
[1] What is Mass Spectrometry? Ed. Am. Soc. Mass Spectrom., (1998)
[2] (a) Liebler, D. C. Introduction to Proteomics- Tools for the New Biology,
Human Press, Totowa, NJ, 2002; (b) Mann, M. y Jensen, O. N. Proteomic analysis of post-translational modifications (www.nature.com/naturebiotechnology) Vol 21 March 2003; Werner, T. Proteomics and Regulomics: The Yin and Yang of Functinal Genomics, Mass Spectrom. Rev., 23, 2004, 25-33.
[3] Siuzdak, Mass Spectrometry for Biotechnology, Academic Press (1996).
[4] (a) Dell, A. y Morris, H. Glycoprotein Structure Determination by Mass Spectrometry. Science 291, 2001, 2351-2356. (b) A Hlenius, A. y Aebi, M.
Intracellular functions of N-lynked Glycans. Science 291, 2001, 2364-2369.
[5] Thompson, J. J., Philos. Mag., 20, 1910, 752.
[6] McLafferty, F. W. and Turecek, F. Interpretation of mass spectra,
4th ed., California, University Science Books, 1993.
[7] Back to Basics Manual, Micromass UK Limited, www.micromass.co.uk
[8] Budzikiewicz, H., Djerassi, C. and Williams, D. H., Mass spectrometry of organic compouds. San Francisco: Holden-Day, 1967.
[9] Budzikiewicz, H., Djerassi, C. and Williams, D. H., Structure elucidation of natural products by mass spectrometry.
San Francisco : Holden-Day, 1964.
[10]M. Barber and B. N. Green, Rapid Comunn. Mass Spectrom., 1, 1987, 80-83
[11]A. Benninghoven, Angew. Chem. Int. Ed. Engl., 33, 1994, 1023-1043.
[12]R. D. Mcfarlane and D. F. Torgerson, Science, 191, 1976, 920; B. Sundqvist and R. D. Mcfarlane, Mass Spectrom. Rev. 4, 1985, 421-460.
[13] F. J. Vastola, O. Mumma and A. J. Pirone, J. Org. Mass Spectrom., 3, 1970, 101; M. A. Posthumus, P. G. Kistemaker, H. L. C. Meuzelaar and M. C. Ten Noever de Brauw, Anal. Chem., 50, 1978, 985.
[14] J. Org. Mass Spectrom., ahora se llama J. Mass Spectrom.
[15] Cotter, R. J. Anal. Chim. Acta, 195, 1987, 45-59.
[16] Tanaka, K., Ido, Y., Akita, S., Yoshida, Y., Yoshida, T. Detection of high mass molecules by laser desorption time-of-flight mass spectrometry. Proceed. 2nd Japan-China Joint Symp. Mass Spectrom., pp.185-188 (1987).
[17] Tanaka, K., The origin of macromolecule ionization by laser irradiation, Nobel Lecture, http//www.nobel.se
[18] Tanaka, K., Waki, H., Ido, Y., Akita, S., Yoshida, Y., Yoshida, T. Protein and polymer analyses up to m/z 100 000 by Laser Ioniation Time-of-Flight Mass spectrometry. Rapid Comunn. Mass Spectrom., 2, 1988, 151-153.
[19] Karas, M. and Hillenkamp, F. Laser desorption Ionization of proteins with molecular masses exceeding 10 000 Daltons, Anal. Chem., 60, 1988, 2299-2301.
[20] Cozzolino, R., Passalacqua, S., Salemi, S., Malvagna, P., Spina, E. y Garozzo, D. Identification of adulteration in milk by matrix-assited laser deaorption/ionization time-of-flight mass spectrometry. J. Mass Spectrom., 36, 2001, 1031-1037.
[21] Todd, P. J., Gregory Schaaff, T., Chaurand, P. y Caprioli, R. M. Organic ion imaging of biological tissue with secondary ion mass spectrometry and matrix-assisted laser desorption/ionization. J. Mass Spectrom. 36, 2001, 355–369.
[22] Zaia, J. Mass spectrometry of oligosaccharides. Mass Spectrom. Rev., 23, 2004, 161-227.
[23] (a) Nonami, H., Fukui, S. y Erra-Balsells, R. b-Carboline Alkaloids as Matrices for Matrix-assisted Ultraviolet Laser Desorption Time-of-flight Mass Spectrometry of Proteins and Sulfated Oligosaccharides: A Comparative Study Using
Phenylcarbonyl Compounds, Carbazoles and Classical Matrices. J. Mass Spectrom., 32, 1997, 287-296. (b) Nonami, H., Tanaka, K., Fukuyama, Y. y Erra-Balsells, R. b-Carboline Alkaloids as Matrices for UV-Matrix-assisted Laser Desorption/Ionization Time-of-flight Mass Spectrometry in Positive and Negative Ion Modes. Analysis of Proteins of High Molecular Mass, and of Cyclic and Acyclic Oligosaccharides. Rapid Commun. Mass Spectrom., 12, 1998, 285-296. (c) Nonami, H., Wu, F., Thummel, R. P., Fukuyama, Y., Yamaoka, H. y Erra-Balsells, R. Evaluation of Pyridoindoles,
Pyridylindoles and Pyridylpyridoindoles as Matrices for UV-Matrix-assisted Laser
Desorption/Ionization Time-of-flight Mass Spectrometry. Rapid Commun. Mass Spectrom., 15, 2001, 2354-2373. (d) Erra-Balsells, R y Nonami, H. Nor-Harmane (9H-Pyrido[3,4-b]indole) as Outstanding Matrix for UV-Matrix-assisted Laser Desorption /Ionization Time-of-flight Mass Spectrometry Analysis of Synthetic and Bio-Polymers, Environ. Control in Biol., 40, 2002, 55-73,
ISSN 1666-7948
www.quimicaviva.qb.fcen.uba.ar